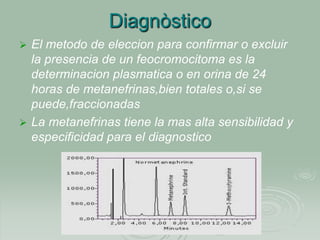
Este documento describe el feocromocitoma, un tumor poco frecuente que se asienta principalmente en la médula suprarrenal y produce cantidades excesivas de catecolaminas. Explica que la mayoría de los feocromocitomas son benignos, pero un 10% son malignos. También cubre la etiología, fisiopatología, síntomas clínicos, manifestaciones metabólicas, diagnóstico y tratamiento de esta condición.