Descargado 2031 veces

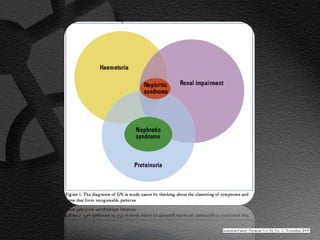



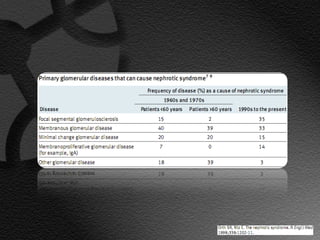
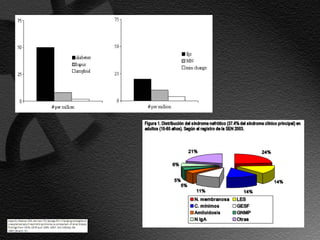


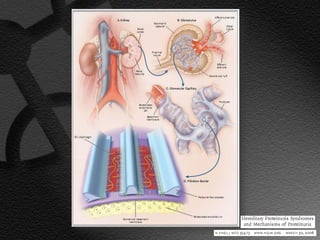

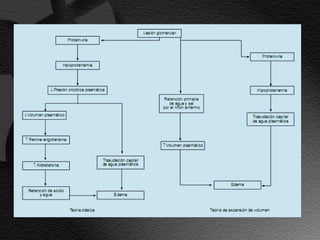



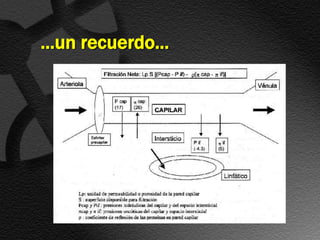




















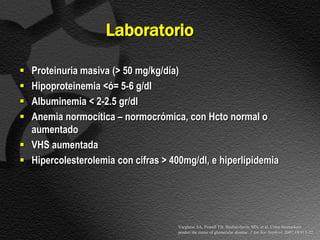













Este documento describe el síndrome nefrótico, incluyendo su definición, etiología, fisiopatología, manifestaciones clínicas y exámenes de laboratorio. El síndrome nefrótico se caracteriza por proteinuria masiva, hipoalbuminemia e hiperlipidemia, y puede ser causado por una variedad de enfermedades renales. Su mecanismo central es un aumento de la permeabilidad glomerular que conduce a la pérdida de proteínas en la orina.




































































































