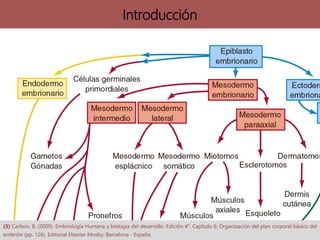
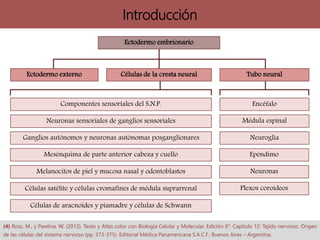
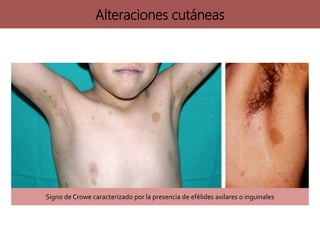
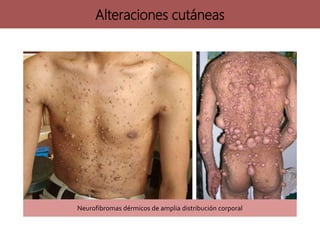
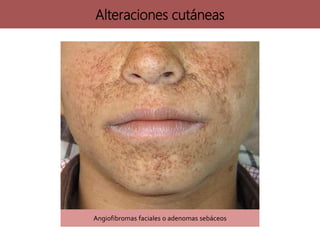
Los síndromes neurocutáneos son afecciones hereditarias caracterizadas por alteraciones en estructuras ectodérmicas que afectan la piel y el sistema nervioso, incluyendo condiciones como neurofibromatosis y esclerosis tuberosa. Estas enfermedades se clasifican según su herencia genética y presentan diversas manifestaciones clínicas que incluyen lesiones cutáneas y alteraciones neurológicas. El diagnóstico se basa en criterios clínicos específicos y en la identificación de síntomas asociados.






![Presentación
44%
17%
14%
8%
6%
5%
3% 3%
Neurofibromatosis tipo 1
Escrerosis tuberosa
Síndrome de Sturge-Weber
Neurofibromatosis tipo 2
Melanosis cutánea
Xeroderma pigmentoso
Síndrome de Klippel-Trenaunay
Síndrome de Parry-Romberg
(6) R.M. Durón, et al. 2009. Síndromes neurocutáneos en la consulta neurológica. Serie de casos. Rev Med Hondur 2009;77(4): 153-
192. [Revisado noviembre 12, 2015]. Disponible en línea: http://www.bvs.hn/RMH/pdf/2009/pdf/Vol77-4-2009-7.pdf](https://image.slidesharecdn.com/sndromesneurocutneos-151118202516-lva1-app6892/85/Sindromes-neurocutaneos-8-320.jpg)
![Presentación
35%
22%
22%
13%
5% 3%
Control neurológico
Cefalea
Epilepsia
Déficit motor
Déficit visual
Radiculopatías
(6) R.M. Durón, et al. 2009. Síndromes neurocutáneos en la consulta neurológica. Serie de casos. Rev Med Hondur 2009;77(4): 153-
192. [Revisado noviembre 12, 2015]. Disponible en línea: http://www.bvs.hn/RMH/pdf/2009/pdf/Vol77-4-2009-7.pdf](https://image.slidesharecdn.com/sndromesneurocutneos-151118202516-lva1-app6892/85/Sindromes-neurocutaneos-9-320.jpg)





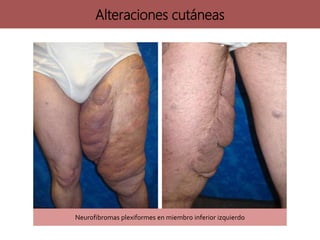
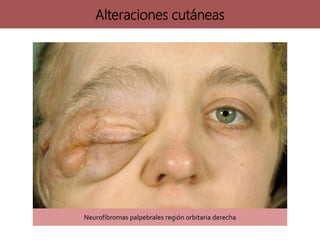






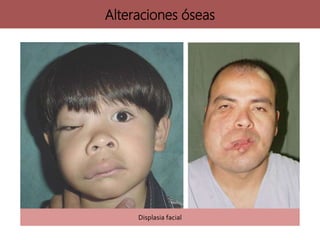






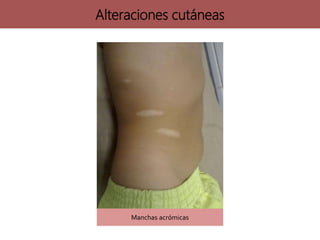


















































































![neurofibromatosis[1] TIPOS DE NEUROFIBROMATOSIS](https://cdn.slidesharecdn.com/ss_thumbnails/neurofibromatosis1-250703211636-b811cee8-thumbnail.jpg?width=640&height=640&fit=bounds)





























