



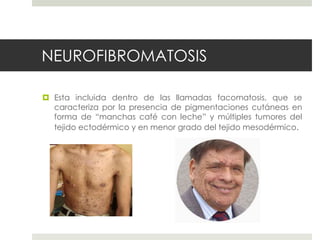




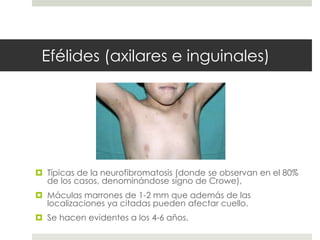
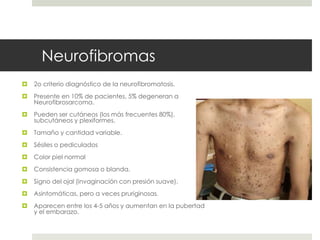
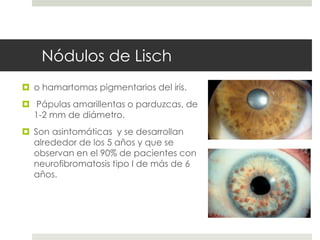





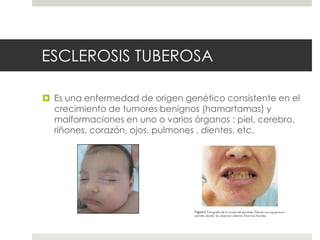


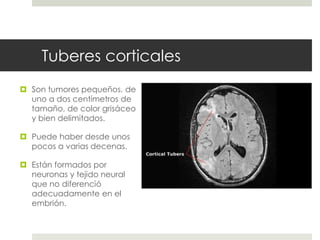
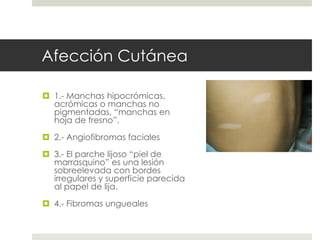
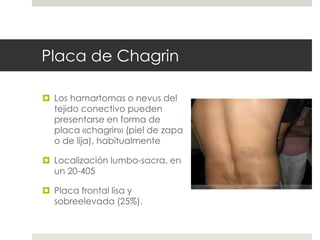
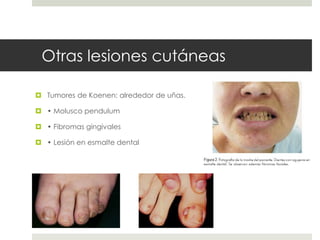

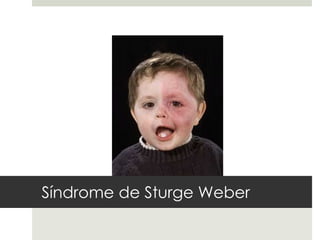
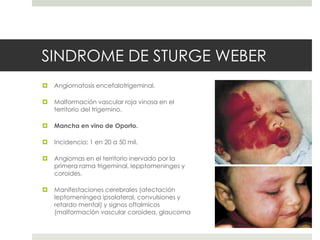



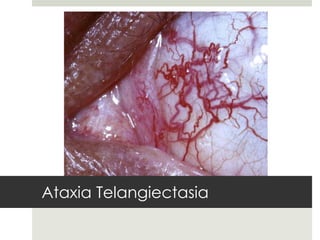




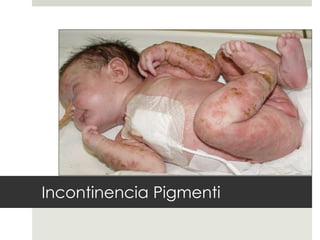


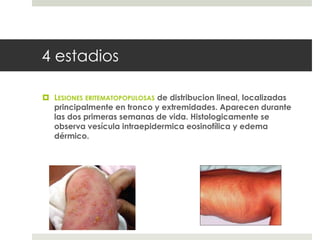


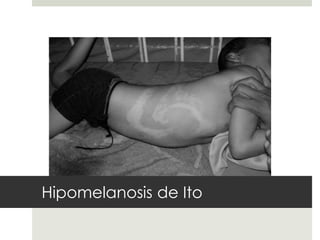


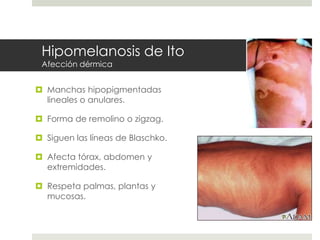
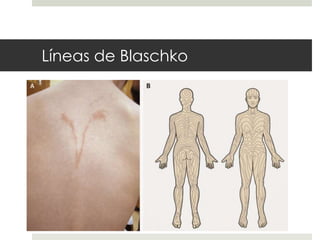
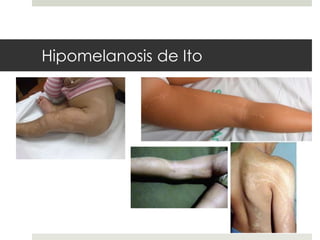
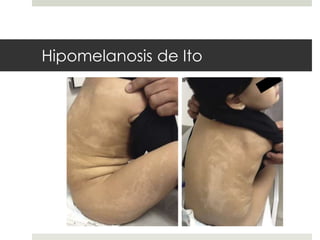
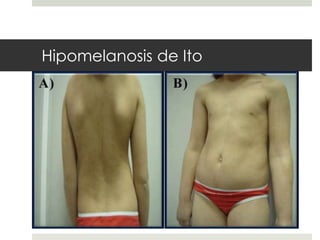
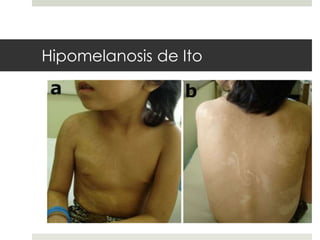
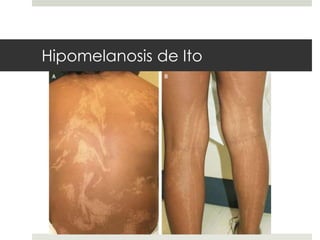
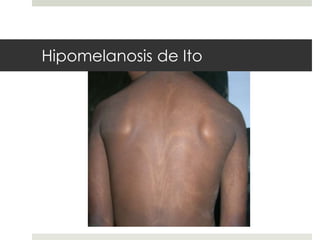
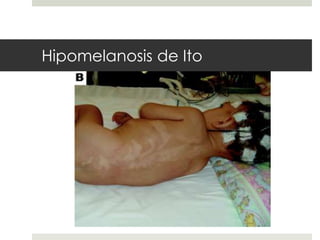
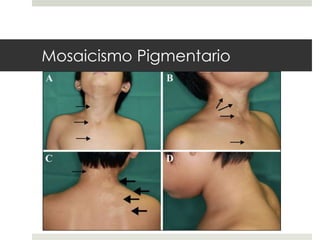






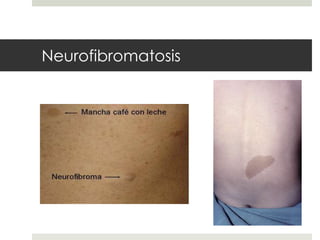
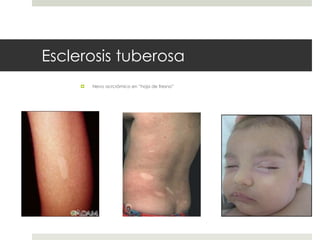
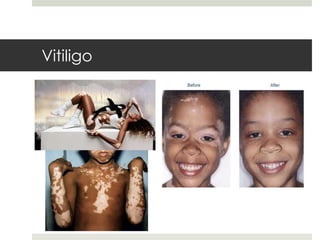
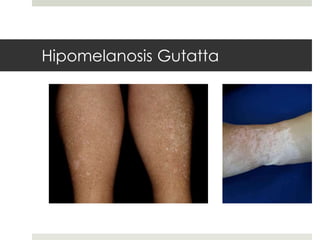
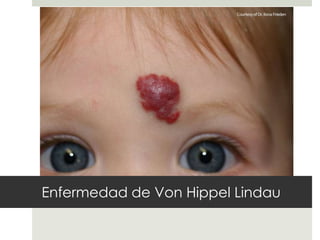




Este documento describe varios síndromes neurocutáneos, incluyendo la neurofibromatosis, la esclerosis tuberosa, el síndrome de Sturge Weber, la ataxia telangiectasia, la incontinencia pigmenti y sus principales características clínicas. Estos síndromes se caracterizan por afectar tanto la piel como el sistema nervioso central y periférico, y se heredan de forma autosómica dominante o recesiva. Cada uno presenta manifestaciones dermatológicas, neurológicas y oftalmológic










































































