Descargado 278 veces




























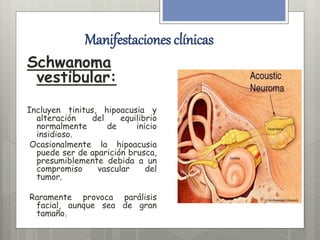








![MUTACIONES MÁS FRECUENTES ESCLEROSIS TUBEROSA
Gen % de mutaciones en Método
Frecuencia de la mutación
Caso familiar Caso único
mutations were missense, nonsense, and a one-nucleotide
insertion or deletion.
TSC1 ~31%
Secuenciación ~30% ~15%
Análisis de
Deleción
0 ~1%
/duplicación
TSC2 ~69%
Secuenciación
50% ~60%-70%
Análisis de
Deleción
/duplicación
~1% ~6%
GeneReviews® [Internet].](https://image.slidesharecdn.com/facomatosisexpo-141117104837-conversion-gate01/85/Facomatosis-42-320.jpg)











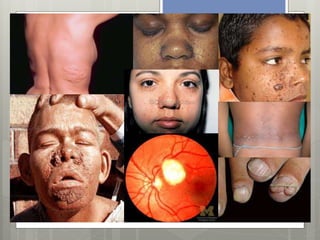

















































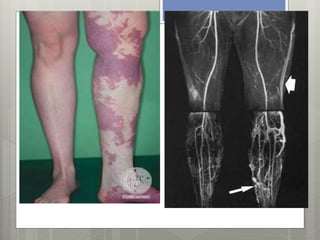
![Bibliografía:
Fernandez Concepción, Gomez García et al., Síndrome de
Sturge Weber. Revisión. Rev CubanaPediatr 1999;71(3):153-
9.
Convusions and homonymous hemianopsia as initial manif
estations of Sturge-Weber syndrome in a 64-year-old male
. García-Estévez DA. Neurologia. 2013 Mar 2. doi:pii: S0213-
4853(13)00010-8. 10.1016/j.nrl.2013.01.004. [Epub ahead of
print] English, Spanish.
http://www.omim.org/](https://image.slidesharecdn.com/facomatosisexpo-141117104837-conversion-gate01/85/Facomatosis-107-320.jpg)

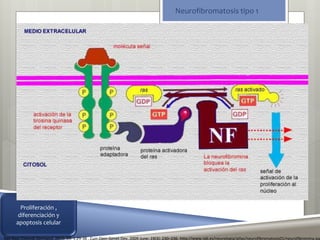
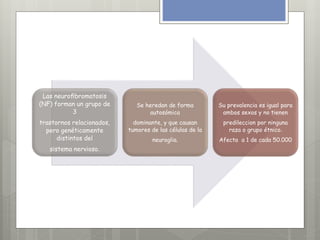
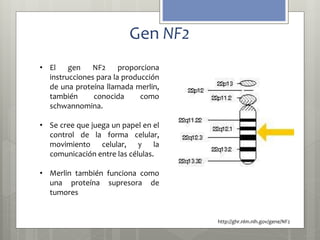
El documento trata sobre la neurofibromatosis tipo 1. En primer lugar, define la neurofibromatosis como un grupo heterogéneo de síndromes genéticos que se caracterizan por afectar múltiples sistemas y causar lesiones cutáneas y tumorales del sistema nervioso. Luego, describe las principales características clínicas y genéticas de la neurofibromatosis tipo 1, incluyendo su herencia autosómica dominante y las mutaciones en el gen NF1 que codifica la proteína neurofibromina. Finalmente, explica los criter

















































![neurofibromatosis[1] TIPOS DE NEUROFIBROMATOSIS](https://cdn.slidesharecdn.com/ss_thumbnails/neurofibromatosis1-250703211636-b811cee8-thumbnail.jpg?width=640&height=640&fit=bounds)














































