Descargado 31 veces


















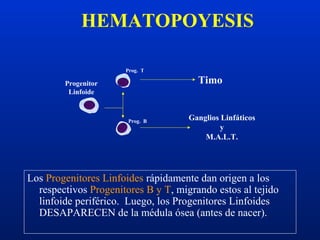
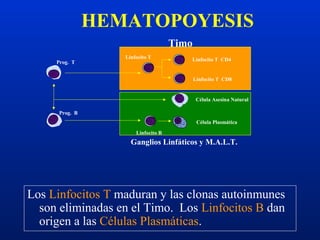


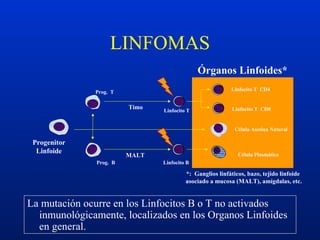

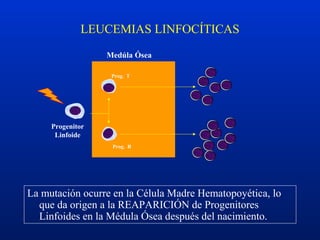
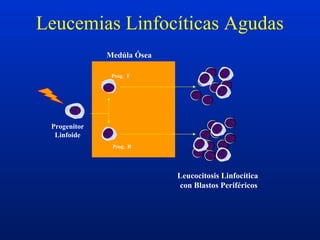



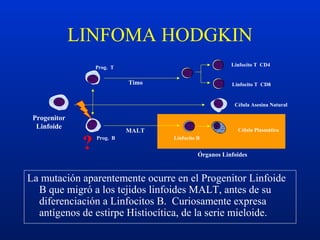



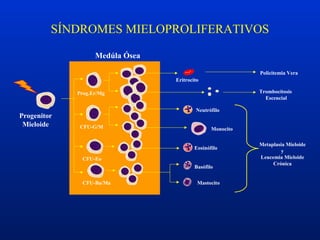

Este documento resume diferentes trastornos hematológicos. Describe las anemias, incluyendo las causadas por pérdida de sangre, hemolíticas y por reducción en la producción de eritrocitos. También cubre la policitemia y las neoplasias hematológicas como las leucemias, linfomas y síndromes mieloproliferativos.



































































































