


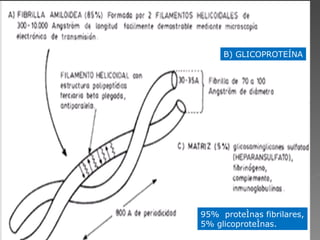






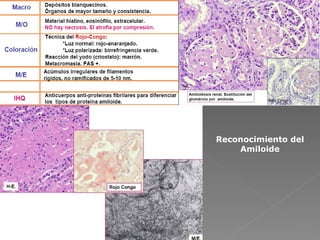



































La amiloidosis es una enfermedad caracterizada por el depósito extracelular de proteínas fibrilares llamadas amiloide en diversos órganos. Puede ser primaria o secundaria a otras enfermedades. Se diagnostica mediante biopsia y tinción de los tejidos con colorantes específicos. Los órganos más frecuentemente afectados son el riñón, corazón e hígado, pudiendo causar insuficiencia renal, arritmias e insuficiencia hepática.























































































