Descargado 214 veces



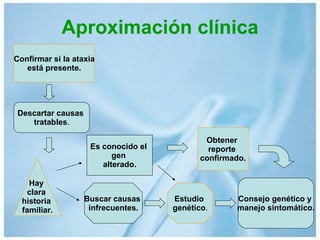






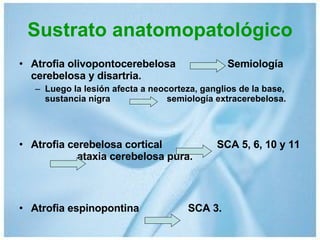

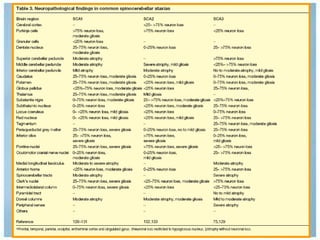


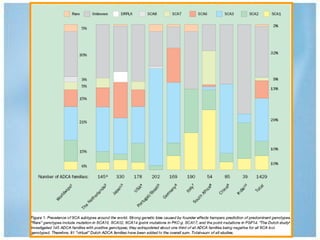
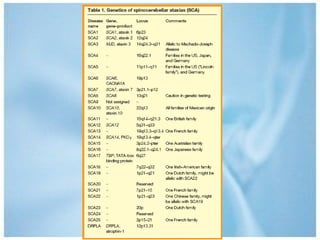
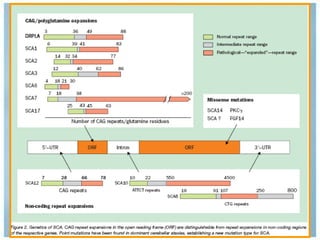
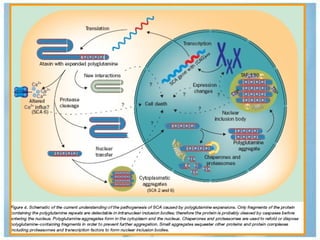
















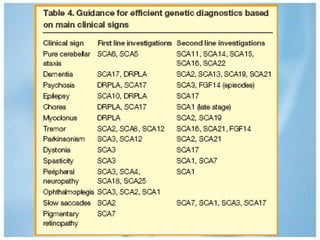


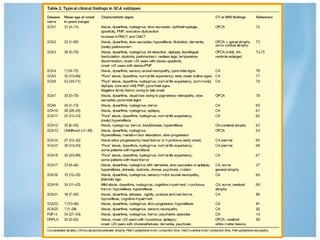



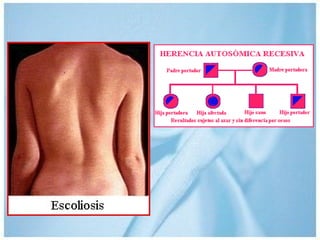
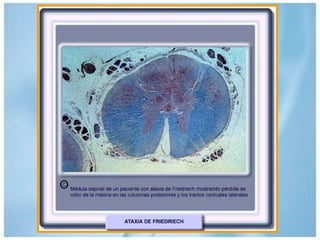


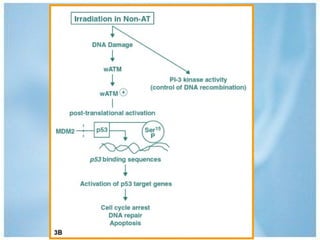







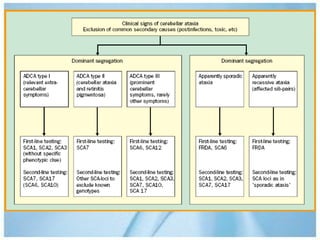



Las ataxias hereditarias son un grupo de desórdenes neurodegenerativos caracterizados por incoordinación y pérdida del balance que conducen a discapacidad. Incluyen ataxias autosómicas dominantes causadas por la expansión de tripletes en diferentes genes, ataxias autosómicas recesivas como la de Friedreich ligada a la expansión GAA, y ataxias ligadas al cromosoma X como la telangectasia. El diagnóstico requiere descartar causas adquiridas y estudiar el patrón de herencia cuando existe histor



































































