Descargado 377 veces







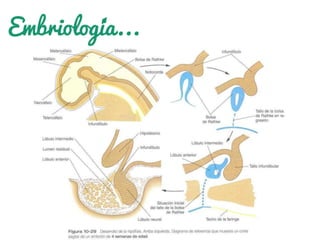


















Los craneofaringiomas son tumores benignos y de lento crecimiento que se localizan en las regiones selar y paraselar del sistema nervioso central. Representan entre el 3% y 10% de las neoplasias intracraneales. Se observan con más frecuencia en la infancia y adolescencia. Los principales síntomas son defectos visuales, hipertensión intracraneal y alteraciones en la función de la glándula pituitaria. El tratamiento ideal es la resección quirúrgica total.










































































