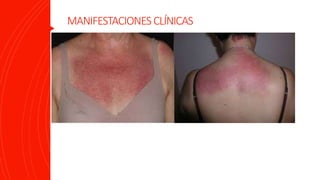
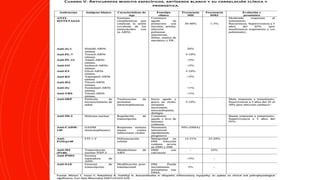
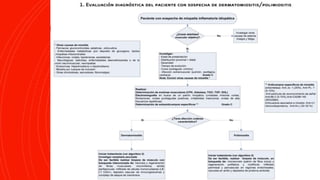
La dermatomiositis es una miopatía inflamatoria autoinmune caracterizada por debilidad muscular, inflamación muscular, manifestaciones cutáneas como pápulas de Gottron y eritema heliotropo, y afectación de otros órganos. Su diagnóstico requiere evaluación clínica, histopatológica y serológica. El tratamiento consiste principalmente en corticosteroides e inmunosupresores para controlar la inflamación muscular.