
Descargado 23 veces

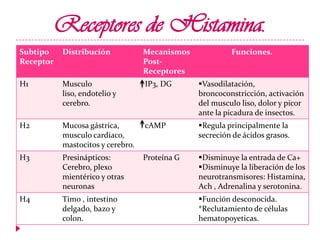
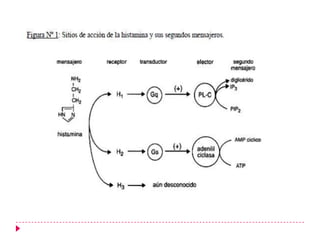

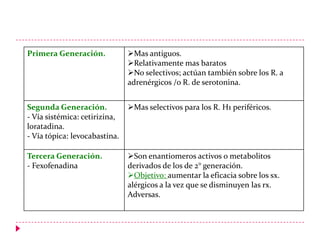


























Este documento presenta información sobre diferentes medicamentos utilizados para tratar la rinitis alérgica, rinitis vasomotora, urticaria y edema vasogénico. Explica los receptores de histamina, antihistamínicos de primera, segunda y tercera generación como la clorfenamina, loratadina y fexofenadina, así como esteroides y sus efectos.























































































