Descargar como PDF, PPTX






















































![Bibliografía
Jones, K., Jones, M. and Campo, M. (n.d.). Smith's recognizable patterns of
human malformation.
Omim.org. (2016). OMIM - Online Mendelian Inheritance in Man. [online] Available
at: http://www.omim.org
Sx Pallister Killian:
Novel Clinical manifestations in Pallister-Killian Syndrome: Comprehensive Evaluation onf 59 affected
individuals and review of previous reported cases
http://www.pkskids.net/documents/WilkensetalPKSClinicalOverviewAJMG12-12.pdf
Sx Cowden:
Queratosis acras y queratosis folicular invertida como manifestación de la enfermedad de Cowden
http://actasdermo.org/es/queratosis-acras-queratosis-folicular-invertida/articulo/13107645/](https://image.slidesharecdn.com/fenotiposmodulo10-161114010612/85/Fenotipos-modulo-10-58-320.jpg)

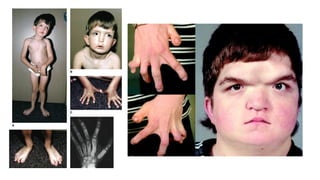
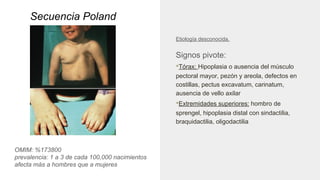
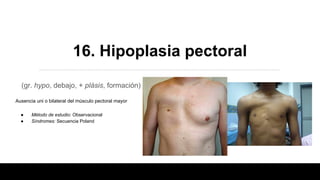
Este documento describe 28 fenotipos relacionados con el cuello, tronco y mamas. Se proporciona información sobre el método de estudio y síndromes asociados con cada fenotipo. Algunos de los fenotipos descritos incluyen pterigium colli, piel redundante en el cuello, cuello corto, clavículas ausentes, hombro de Sprengel, ginecomastia, pezones prominentes y politelia. Para cada fenotipo, se especifican los síndromes asociados y otros detal





















































































