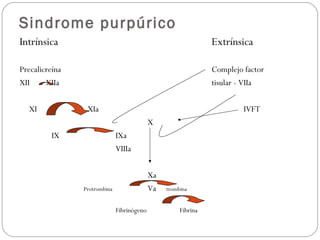
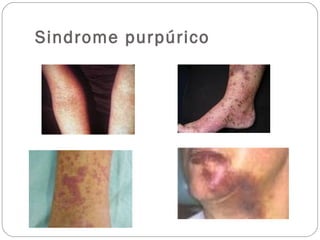
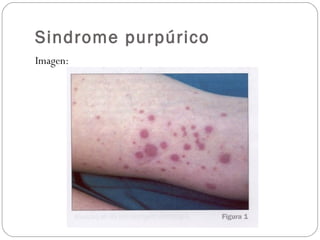
El documento describe el síndrome purpúrico y los mecanismos de hemostasia primaria y secundaria. Explica cómo evaluar a pacientes con síndrome purpúrico mediante el interrogatorio, examen físico y pruebas de laboratorio como recuento de plaquetas, tiempo de sangrado, TP y TTPa. También presenta dos casos clínicos de pacientes con síndrome purpúrico, uno diagnosticado con púrpura trombocitopénica inmune y el otro con vasculitis leucocitoclástica sec