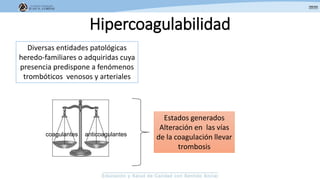
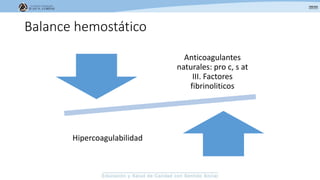
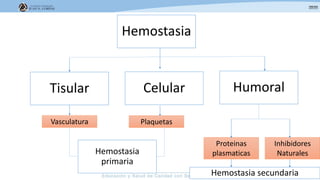
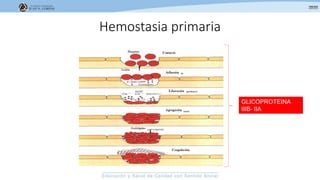
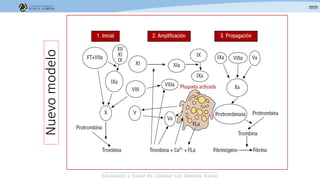
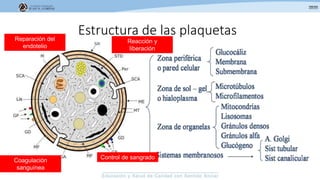
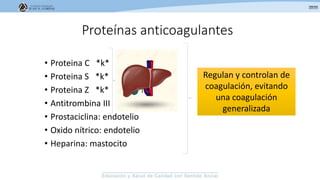
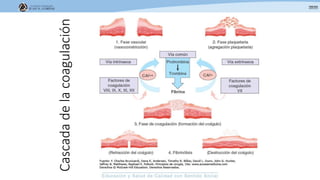
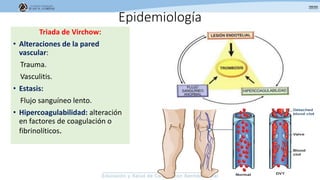
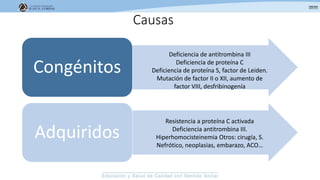
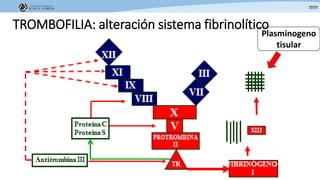
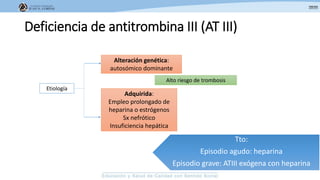
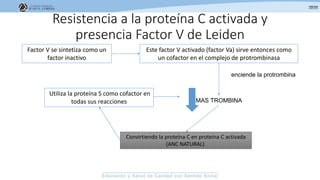
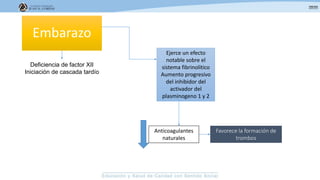
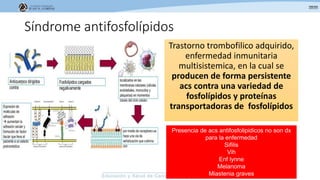
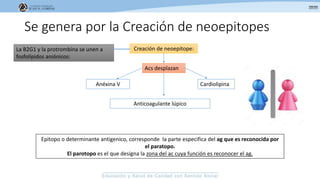
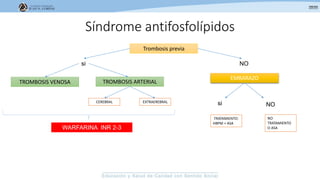
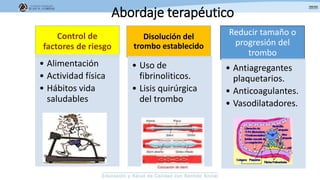
Los síndromes trombóticos se refieren a estados de hipercoagulabilidad causados por alteraciones en las vías de la coagulación que predisponen a fenómenos trombóticos. Estos pueden ser hereditarios o adquiridos y se deben a deficiencias en proteínas anticoagulantes o aumentos en factores procoagulantes. El diagnóstico y tratamiento adecuados dependen de identificar la causa subyacente para prevenir eventos trombóticos futuros mediante el uso de anticoagulantes.