Descargado 108 veces

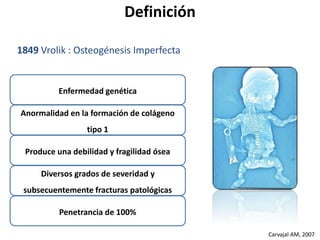
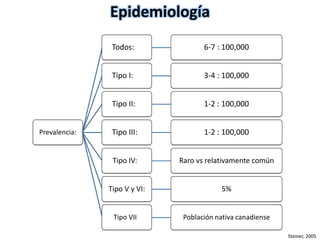

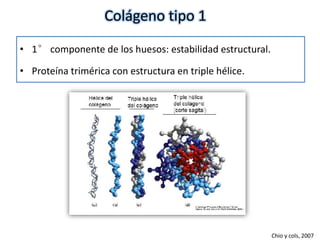
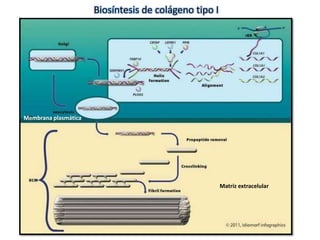
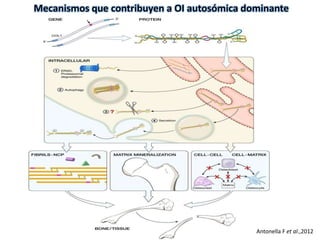
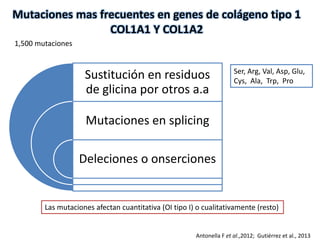
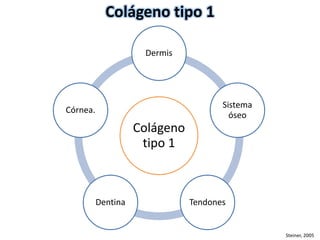
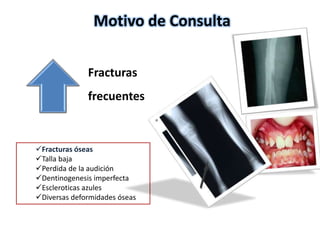
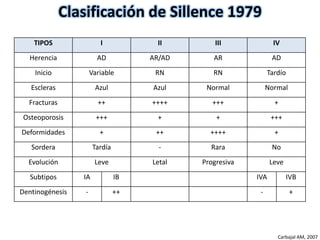
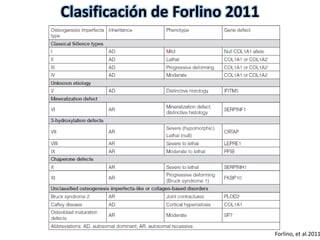
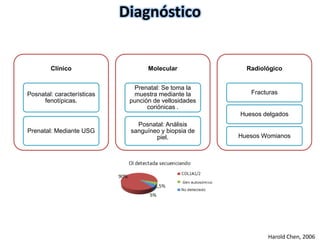
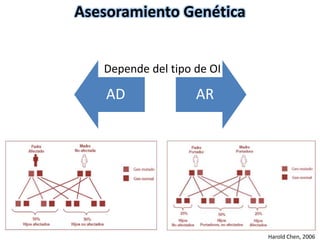

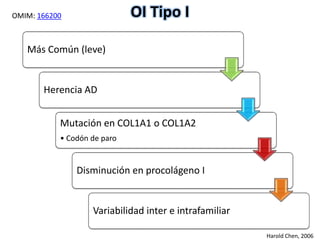
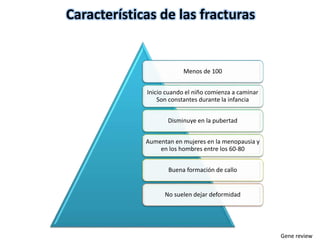
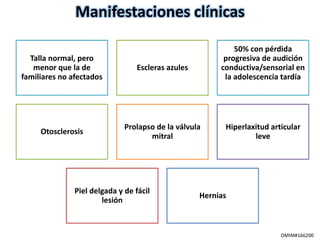
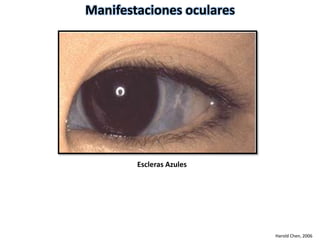
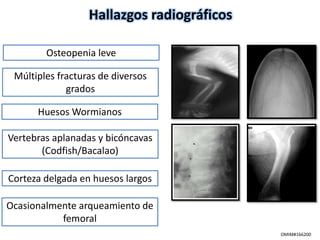
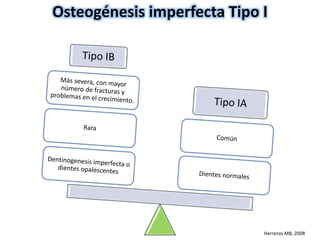


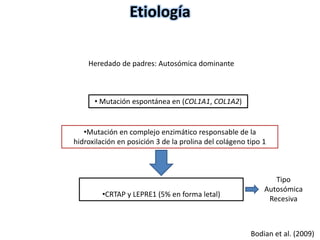
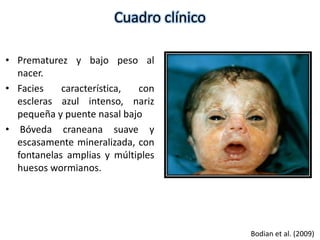
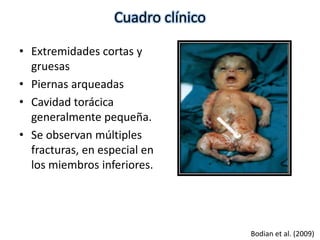

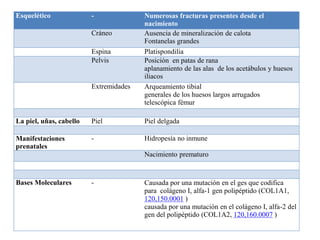
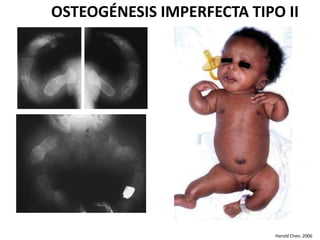
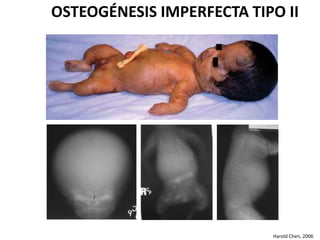
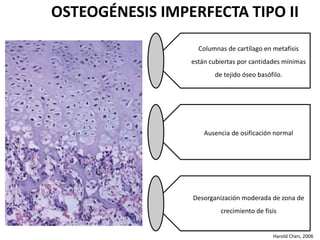
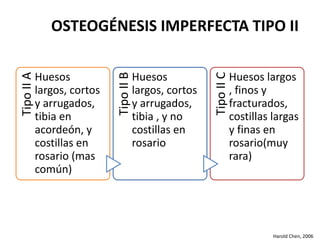
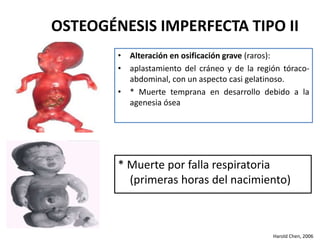


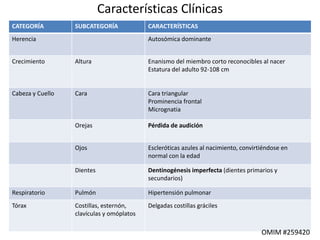
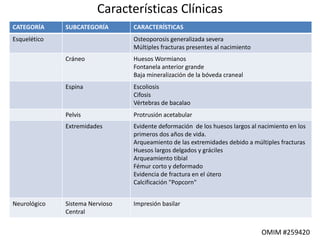
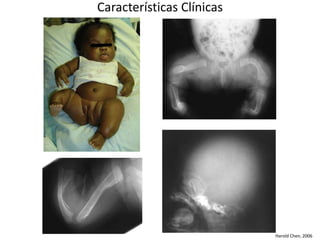
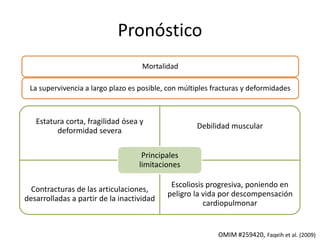


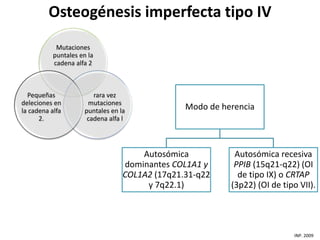
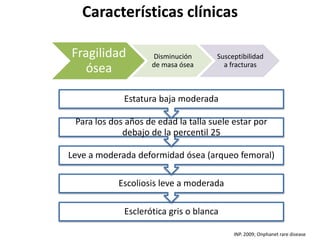


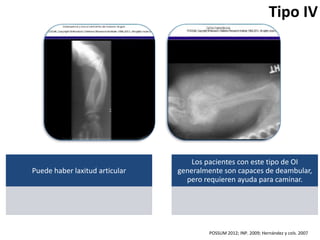

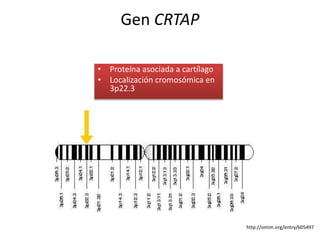

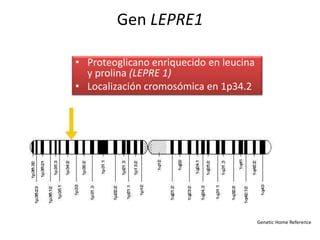

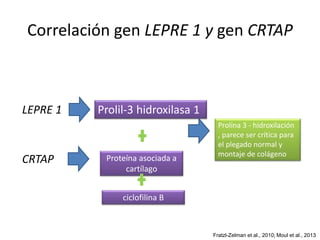
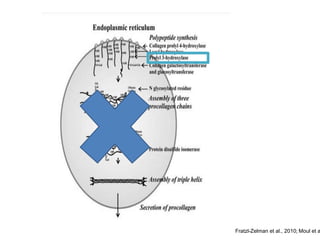

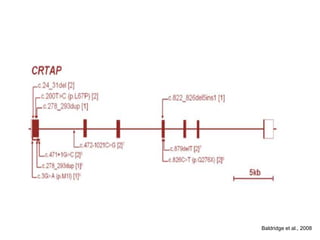
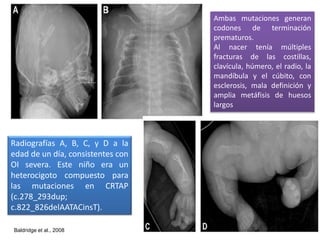
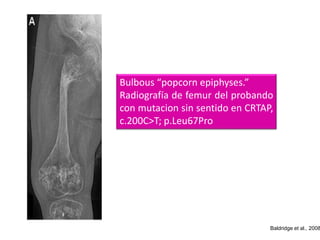

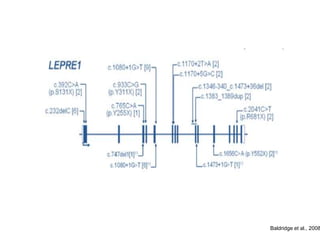
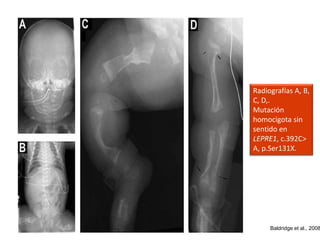

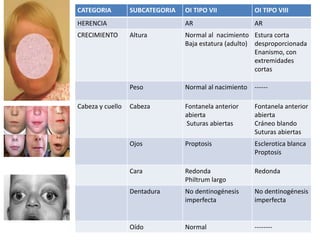
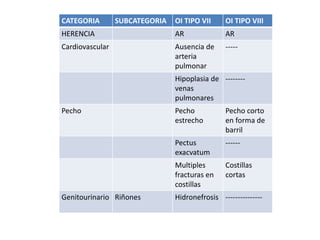
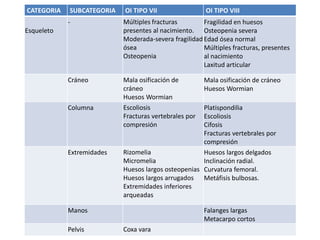
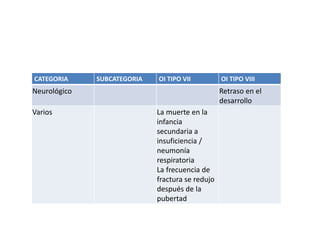



Este documento resume los tipos de osteogénesis imperfecta (OI), una enfermedad genética que causa fragilidad ósea. Describe los cuatro principales tipos de OI (I-IV), incluidas sus características clínicas, genéticas y radiológicas. El tipo I es el más común y leve, mientras que el tipo II es mortal en el período prenatal. El tipo III causa deformidades óseas progresivas y el tipo IV es la forma más variable con fragilidad ósea moderada.




















































![Enfermedades oseas generalidades.dr elvisac [modo de compatibilidad]](https://cdn.slidesharecdn.com/ss_thumbnails/enfermedadesoseasgeneralidades-drelvisacmododecompatibilidad-110428205237-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



















































