

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Acondroplasia
Similar a Acondroplasia (20)
Más de Oswaldo A. Garibay
Más de Oswaldo A. Garibay (20)
Último
Último (20)
Acondroplasia
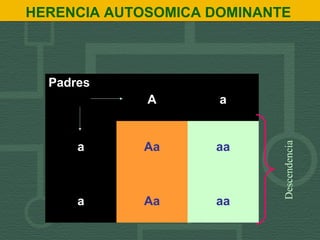
- 1. HERENCIA AUTOSOMICA DOMINANTE Padres A a a Aa aa a Aa aa Descendencia
- 2. HOMOCIGOTOS AD (gravemente afectados) Padres A a A AA Aa a Aa aa Descendencia
- 4. HERENCIA AUTOSÓMICA DOMINANTE • Síndrome de MarfanSíndrome de Marfan • AcondroplasiaAcondroplasia • Síndrome de Ehlers-DanlosSíndrome de Ehlers-Danlos • Neurofibromatosis (I y II)Neurofibromatosis (I y II)
- 5. SINDROME DE MARFAN • AD • MIM 154700 • Tejido conectivo • Efectos sistémicos • Cardiovasculares • Musculoesqueléticas • Oftalmológicas
- 6. • 2-3/10, 000 • 1/4 – 1/3 mutaciones de novo • Alta penetrancia, expresión variable • Complicaciones cardiovasculares
- 7. DIAGNÓSTICO • CRITERIOS DE GHENT: • Primer caso en la familia: dos criterios mayores en dos órganos y uno menor en otro órgano o sistema • Si se encuentra mutación de FBN1 es necesario un criterio mayor de un sistema y afectación de otro • En un familiar de un paciente con dx confirmado de Sx de Marfan es necesario un criterio mayor de un sistema y afectación de otro
- 8. SISTEMA ESQUELÉTICO • Requiere por lo menos 2 criterios mayores o 1 mayor mas dos menores • Talla alta • Dolicosestenomelia • Aracnodactilia • Criterio mayor se requieren por lo menos cuatro manifestaciones: – Pectus carinatum o excavatum – RS <1
- 9. – Signo de la muñeca (Walker-murdoch) + si las falanges del 1ero y 2do dedo de una mano se sobreponen al abrazar la muñeca. – Signo del pulgar (Steinberg) + si el pulgar es se opone al cerrar la mano y este sobrepasa el borde cubital – Escoliosis de ≥20º (60%) – Extensión disminuida de codos – Desplazamiento medial del maleolo interno por pie plano – Protusión acetabular (50%)
- 10. CARACTERISTICAS CLINICAS • Alta estatura • Dolicoestenomelia • Aracnodactilia • Deformidad columna y tórax anterior • Hipermobilidad articular • Dilatación y disección de la aorta ascendente • Prolapso de válvula mitral • Miopia • Ectopia lentis
- 20. SISTEMA ESQUELETICO • Pectus carinatum, Pectus excavatum • Signos de la muñeca y los pulgares positivos • Escoliosis severa • Desplazamiento medial del maleolo medial causando pies planos • Relación de segmentos <1 • Hipermobilidad articular • Paladar alto arqueado con alteraciones dentales • Apariencia facial (dolicocefalia, hipoplasia malar, retrognatia)
- 21. ALTERACIONES OFTALMOLOGICAS • Ectopia lentis • Córnea anormalmente plana • Longitud axial del globo ocular incrementada • Iris Hipoplásico o hipoplasia de los músculos filiares que causan disminución de la miosis
- 22. SISTEMA CARDIOVASCULAR • Dilatación de la aorta ascendente sin regurgitación aórtica • Disección de la aorta ascendente • Prolapso de la válvula mitral con o sin regurgitación • Dilatación de la arteria pulmonar principal en ausencia de estenosis valvular antes de los 40 años • Calcificación del anillo mitral antes de los 40 años • Dilatación o disección de la aorta torácica o abdominal antes de los 50 años
- 23. PIEL Y TEGUMENTOS • Estrías atróficas • Hernias recurrentes
- 24. HISTORIA FAMILIAR • Padre, hijo o familiar afectado • Presencia de una mutación en FBNI causante de Marfan • Presencia de un haplotipo cercano a FMNI, heredado por descendiente, asociado sin equivocación con Marfan • Requerimientos para el caso índice
- 25. BIOLOGIA MOLECULAR • Mutaciones en la glicoproteína de matríz extracelular de la fibra elástica: fibrilina • FBN1 15q21.1 • (FBN2- 5q23-31-aracnodactilia cc MIM 121050)
- 26. • Fibrilina: • 350-kDa • Glicoproteína • 1991: FBN1 • 8616 bp • 65 exones • 200 kb
- 27. • Más de 500 mutaciones • 20% causan terminación prematura • 70% missense • Reduce la distensibilidad de la fibra elastica • 24,27,31,32 exones –Mutaciones asociadas a presentación neonatal
- 28. CARACTERÍSTICAS BIOQUIMICAS • Los casos se dividen en 4 grupos: • 1) Síntesis celular al 50% de la cantidad de fibrilina • 2) Síntesis celular en adecuada cantidad de fibrilina pero es secretada menos eficientemente • 3) Síntesis celular y secreción de fibrilina normal pero no se incorporan las moléculas en la matriz extracelular • 4) No se encuentran alteraciones en la síntesis, secreción o incorporación a la matriz extracelular • Estudios familiares muestran que todos los individuos afectados de una misma familiar tienen el mismo defecto
- 29. DIAGNOSTICO DIFERENCIAL Ectopia Lentis AD Mitral valve prolapse Aortic root dilation without dissection Skeletal and Skin abnormalities Síndrome de Shprintzen-Goldberg Aneurisma aórtico familiar Aneurisma aórtico familiar
- 32. • No hay tratamiento curativo • Tratamiento sintomático y preventivo • Oftlamológico • Ortopédico • Cardiovascular
- 33. ACONDROPLASIA
- 35. GENERALIDADES • Acondroplasia (ACH; MIM 100800) es la displasia esquelética más frecuente en humanos. • La incidencia es de 0.5 a 1.5 /1000 RN • En la acondroplasia, la forma mas severa es la displasia tantofórica MIM 187600,y la forma leve es la hipoacondroplasia MIM 146000 todas ellas causadas por mutaciones dominantes del gen FGFR3 que causan activación del Factor de Crecimiento de Fibroblastos tipo 3 (FGFR3)
- 36. HERENCIA • Autosomica Dominante • 80% de los casos son mutaciones de novo • Presencia de edad paterna avanzada en casos esporádicos • Mosaicismo gonadal • Mutaciones en el gen FGFR3 en 4p16.3
- 37. A aA a a aa a A aA a a aa aA aA a a aa a
- 39. • 98% de los casos con acondroplasia presentan una transición G a A y el 1% una transversion G a C en el nucleotido 1138, provocando una substitución de una arginina por glicina en la posición 380 (G380A), del dominio de transmembrana del FGFR3.
- 40. PATOGENESIS • No es bien clara completamente. • FGFR3 se expresa en el anillo de crecimiento epifiseal en las zonas de proliferación. • La activación de FGFR3 cambia el comportamiento de los condroblastos, acortando su periodo de proliferacion y anticipando su diferenciacion a condrocitos.
- 41. • Pérdida cuantitativa de osificación endocondral osificación además de la formación tejido anormal. • Diámetro de los huesos normal secundario a la osificación membranosa por debajo del periostio de huesos tubulares.
- 42. CUADRO CLÍNICO • Alteraciones en la respiración: • 75% durante el sueño • Apnea obstructiva • Enfermedad pulmonar restrictiva
- 43. • Estenosis de medula espinal en mas del 50% de los pacientes: • Cambios de sensibilidad en extremidades inferiores • Incontinencia • Paraplejía • Clasificación de acuerdo a la severidad neurológica y a la estenosis medular
- 44. • TIPO I: dolor en región lumbar, con cambios en la sensibilidad y función motora • TIPO II: claudicación intermitente limitando la ambulación • TIPO III: compresión de raíz nerviosa • TIPO IV: inicio agudo de paraplejía
- 45. ESTENOSIS DEL FORAMEN MAGNO • Dificultad respiratoria • Problemas de succión • Cuadriparesia • Pobre control de la cabeza • Hidrocefalia
- 46. • Talla baja desproporcionada • Hipotonía durante la infancia • Cráneo facial: • Frontal prominente • Puente nasal deprimido • Hipoplasia medio facial • Mal oclusión dental • Estenosis del foramen magno • Estenosis del canal medular
- 48. • Longitud del tronco normal • Toracolumbar xifosis o al nacimiento y/o en la infancia • Hiperlordosis lumbar • Abdomen prominente • Hipermobilidad articular especialmente de rodillas • Acortamiento rizomelico de extremidades
- 50. • Extensión limitada de codo y cadera • Mano en tridente • Braquidactilia
- 51. COMPLICACIONES Y RIESGOS • Otitis media recurrente • Osteo artropatía de la rodilla • Complicaciones neurologicas • Obesidad • Complicaciones obstetricas – DCP – Pelvis estrecha