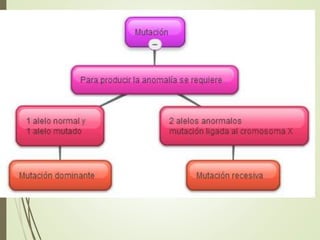
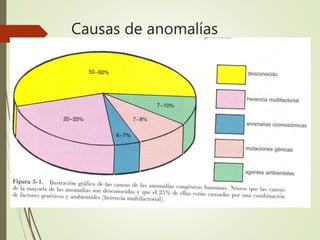
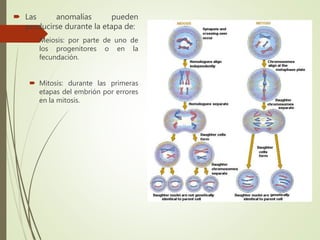
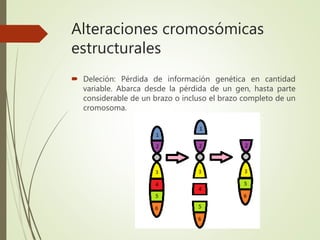
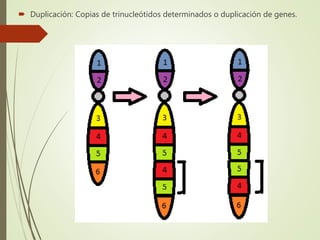
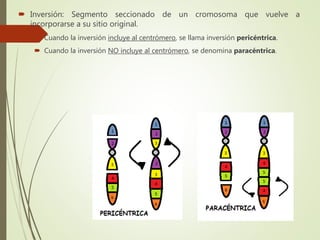
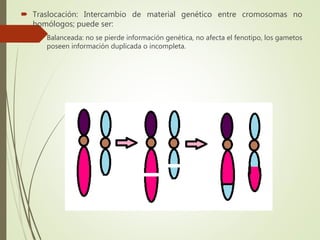
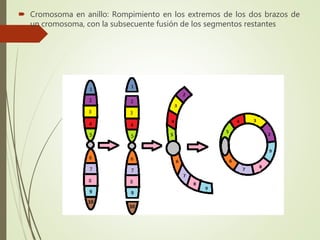
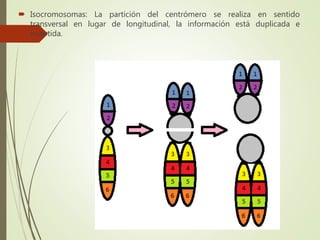
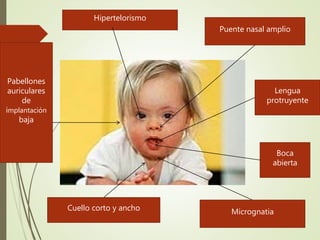
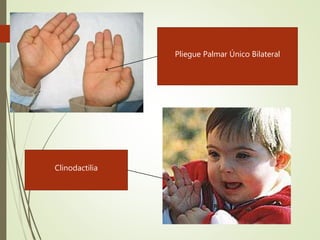
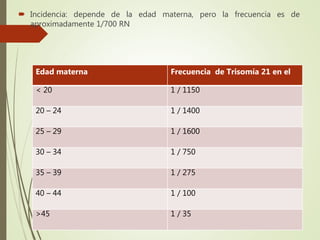
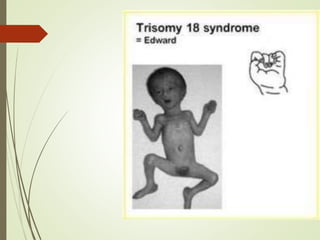
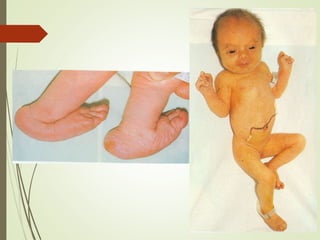
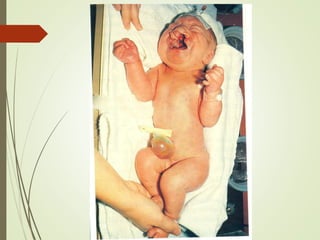
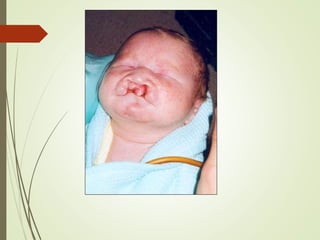
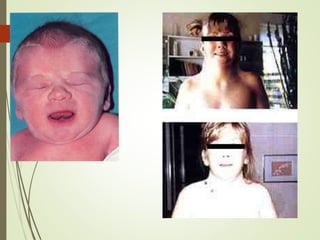
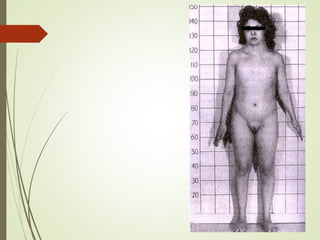
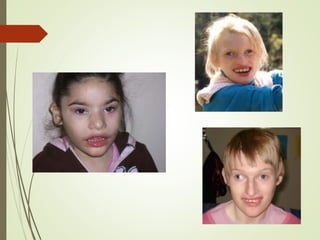
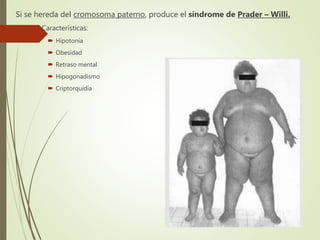
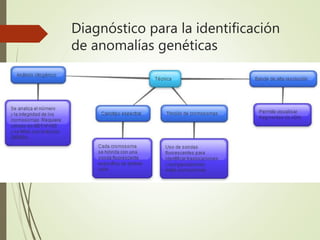
Este documento describe diferentes anomalías y síndromes cromosómicos, incluyendo sus causas y características. Explica que las anomalías pueden ser numéricas, como la trisomía 21 (síndrome de Down), la trisomía 18 (síndrome de Edwards) y la trisomía 13 (síndrome de Patau), o estructurales como deleciones, duplicaciones e inversiones. También describe síndromes como Klinefelter, Turner, triple X, maullido de gato, Prader-Willi, Angelman y otros relacionados