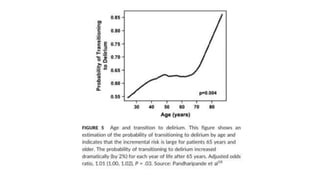
Descargado 17 veces

















![Oxidación: Hipótesis del estrés oxidativo
Especies reactivas de O2 y de
N2 (NO): Estrés oxidativo
daño en la estructura celular
(lípidos, proteínas y DNA).
•Cerebro susceptible por: ↑ [] lípidos, ↑
alta metb estrés oxidativo, ↓ cap
antioxidativa ↑ daño tej cerebral
deterioro cognitivo + degeneración
irreversible Delirium persistente.
Delirium: Expresión
clínica de un defecto
metb cerebral Alt
función cognitiva.
SOH: incap mantener
gradientes ionicos
depresión cortical gralizada,
alt NT, prod radicales libres,
falla en eliminar prod
neurotóxicos.
•IOP: PaO2 < 35mmHg > Riesgo
delirium.](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-20-320.jpg)
![Oxidación: Hipótesis del estrés oxidativo
OSH + NTH: ↓O2 falla metb oxidativo falla bomba ATP: no se
puede mantener gradiente ionico (> influjo Na+ y Ca++, sale K+).
CA++: lib Glutamato (Glu) y Dopamina (DA).
• Glu potencia influjo de Ca++ (q potencia su propia lib): se acumula
en espacio extracell (xq bomba de ATP de glia se encuentra alt)
• DA: ↓ conversión DA NE (O2 dep) y por inhb COMT > [] DA.
• 5HT: ↓ mod corteza, ↑ estriado, estable en tronco.
• ACh: ↓síntesis y lib en basal forebrain ↓ cofactores (tiamina).](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-21-320.jpg)

![Glucocorticoides: Hipótesis Neuroendocrina
• N/ estrés activa Eje Hipotálamo-Hipófisis-
Adrenal (HPA): N. de la base + amígdala
N. paraventricular H: CRH ACTH GC
(cortisol).
• Moviliza depósitos de energía, ↓funciones no
vitales, adaptación fisiológica, mantiene
homeóstasis.
• N/ recuperación estrés: ® GC libres por
retroalimentación (-) involucrando a CPFM,
hipocampo, eje límbico HPA
• Desregulación LHPA: activación crónica ®
GC baja afinidad daño SNC.
• Hipocampo (> [ ] ® GC): > daño.](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-23-320.jpg)
![Glucocorticoides: Hipótesis Neuroendocrina
• Regulación de expresión génica, señalización cell, modulación de
estructuras sinápticas, transmisión y función glial del comportamiento.
• ↑ repetitivo y prolongado de GC alt cap supervivencia neuronal (>
vulnerabilidad) “respuesta aberrante al estres”
Hipótesis Neuroendocrina:
delirium rpta fisiológica por
estrés agudo o crónico por
↑ [] GC](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-24-320.jpg)
![Glucocorticoides: Hipótesis Neuroendocrina
Adultos mayores: > [] cortisol en POD
• Perdida de la función inhibitoria de la esteroideogenesis adrenal Secreción continua en picos de
corticoesteroides.
Cortisol nonsuppression luego de dexametasona: asoc a delirium en 78% con inf TRI.
• ↑ cortisol o desregulación eje LHPA: > riesgo delirium.
↑ GC x enf, trauma, admo externa esteroides ↑ cortisol puede resistencia retroalimentación negativa
por perdida de ® hipocampales.
• GC lib GLU en hipocampo
Uso de corticoesteroides sistémicos.](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-25-320.jpg)

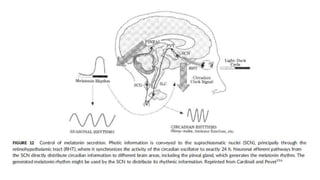

![FACTORES ETIOLOGICOS:
Hipótesis de desconexión de las redes
• Alt balance, función integradora redes neuronales
(usual/ sincronizadas para procesar info sensorial y rpta
motora).
• Hipótesis NDH: cerebro organizado en redes dinámicas
y funcionales anticorrelacionadas.
• ↓ red anticorrelacionada: déficit atencionales.
• Conciencia requiere integridad de red funcional y
conectividades, coperativa pero mutua/ exclusivo paradigma
de introspección (p. ej., red neuronal por defecto [DMN] vs
external awareness (ie, task positive network)](https://image.slidesharecdn.com/delirium-220614014825-aca37090/85/DELIRIUM-pptx-29-320.jpg)







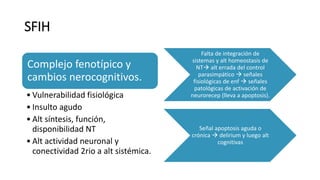



Este documento define el delirium y describe sus diferentes tipos. Explica varias hipótesis sobre los sustratos biológicos que pueden causar delirium, incluyendo la inflamación, el estrés oxidativo, la desregulación del eje hipotálamo-hipófisis-adrenal, la desregulación del ritmo circadiano y la melatonina, y la desconexión de las redes neuronales. También identifica factores de riesgo como la edad y describe cómo los cambios cerebrales relacionados con la edad pueden hacer a los























































































