



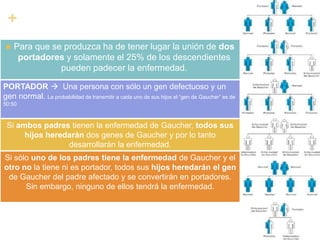





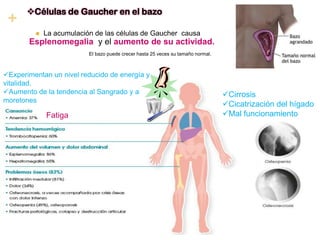



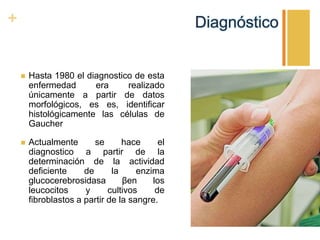





La enfermedad de Gaucher es un trastorno metabólico hereditario causado por la deficiencia de la enzima glucocerebrosidasa, lo que provoca la acumulación anormal de glucocerebroside en los macrófagos. Esto puede causar complicaciones en el hígado, bazo, médula ósea y otros órganos. Actualmente el tratamiento más efectivo es la terapia de reemplazo enzimático, la cual ha mejorado significativamente el pronóstico de los pacientes.


































































![Discromias [Albinismo, Argiria] Enf. Psicosomaticas [Alopecia Areata]](https://cdn.slidesharecdn.com/ss_thumbnails/alopeciaareata-140310151343-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)




















