Descargado 20 veces







![Depuración
La depuración de un fármaco es la tasa de
eliminación por todas las vías
CL= tasa de eliminación/[ ] fármaco
La eliminación no señala la cantidad del
fármaco que se extrae o depura, sino más
bien, el volumen de líquido biológico que
tendría que estar totalmente libre del fármaco
para poder explicar la eliminación](https://image.slidesharecdn.com/fkineticlin-130719125420-phpapp01/85/Fkineticlin-8-320.jpg)

![Depuración
La relación entre la depuración plasmática y
la sanguínea en estado de equilibrio dinámico
está dada por:
CLp = Cb = 1 + H [ Crbc – 1]
CLb Cp Cp
Depuración plasmática total
DPT = Vm/(Km + Cp)
Km, [] plasmática en la cual se llega a la mitad
de la tasa máxima de eliminación (masa/vol)
Vm, es igual a dicha tasa (masa/tiempo)](https://image.slidesharecdn.com/fkineticlin-130719125420-phpapp01/85/Fkineticlin-10-320.jpg)
![Depuración
Para obtener la depuración del fármaco por
parte de un órgano:
Clórgano= Q (CA – CV/ CA)
Donde Q, es el flujo sanguíneo
CA, [] del fármaco en sangre arterial
CV , [] del fármaco en sangre venosa](https://image.slidesharecdn.com/fkineticlin-130719125420-phpapp01/85/Fkineticlin-11-320.jpg)







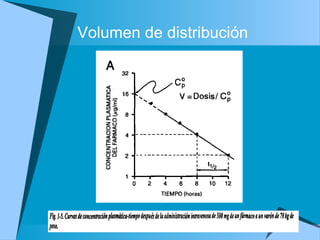
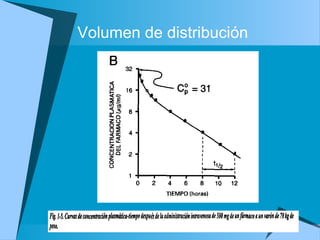



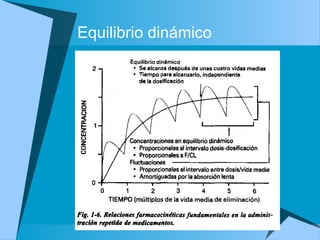





![Dosis de sostén
Dosificación= [] deseada * CL/F
De esta forma se puede calcular la dosis y el
intervalo entre una y otra dosis
La dosis de saturación inicial o dosis de carga
tiene como fin alcanzar pronto la
concentración deseada
Dosis saturación= objetivo deseado Cp * Vss/F](https://image.slidesharecdn.com/fkineticlin-130719125420-phpapp01/85/Fkineticlin-30-320.jpg)

















Este documento introduce conceptos fundamentales de farmacocinética como depuración, volumen de distribución y biodisponibilidad. Explica que la depuración mide la capacidad del cuerpo para eliminar un fármaco, el volumen de distribución representa el espacio donde se distribuye el fármaco, y la biodisponibilidad es la fracción absorbida. También describe cómo estos parámetros se usan para calcular dosis de mantenimiento y de carga inicial para alcanzar concentraciones deseadas de fármacos.



































































