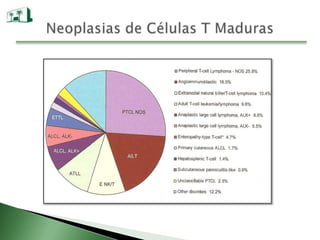
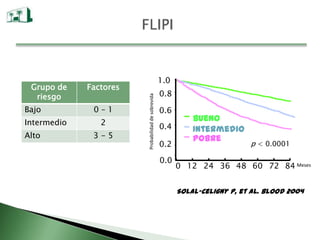
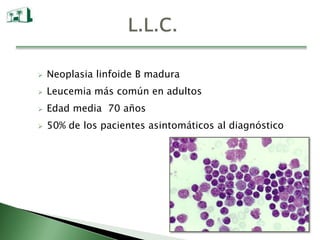
Este documento describe la leucemia linfocítica crónica (LLC), una neoplasia linfocítica B madura. La LLC se caracteriza por la acumulación de linfocitos B clonales en la sangre periférica, médula ósea y ganglios linfáticos. La LLC tiene una presentación variable y su pronóstico depende de factores como el estadio, marcadores biológicos y comorbilidades del paciente. El tratamiento de la LLC también varía dependiendo del grupo de riesgo del paciente.




























































![C:\fakepath\leucemia[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cfakepathleucemia1-100819231029-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)































































