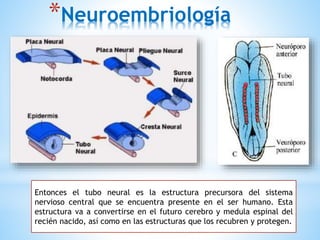
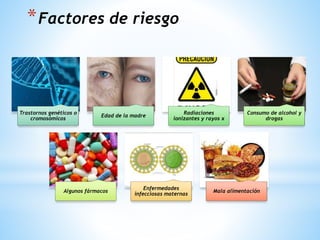
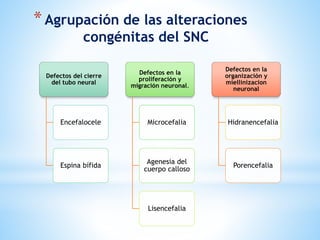
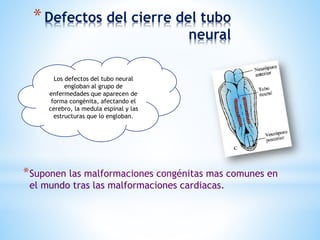
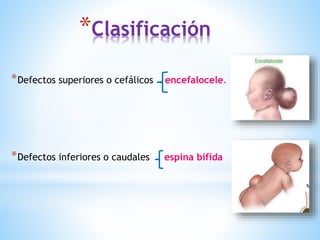
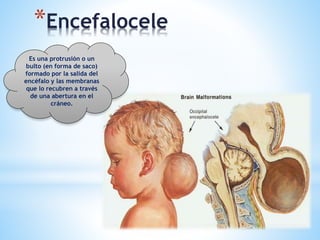
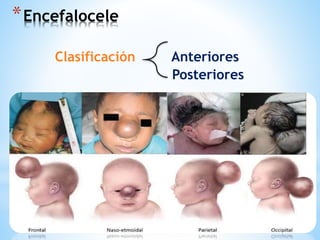
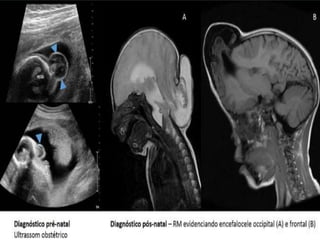
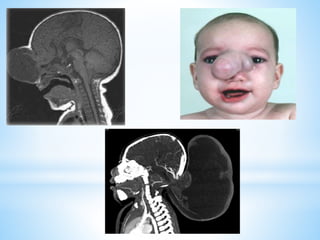
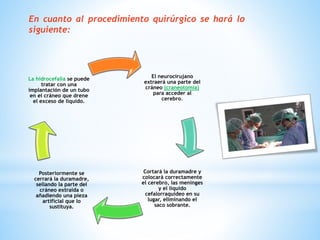
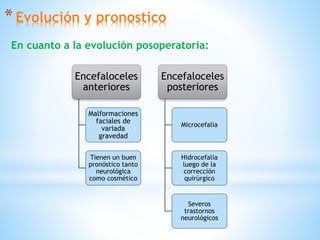
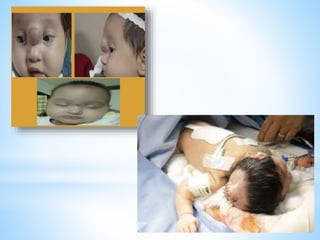
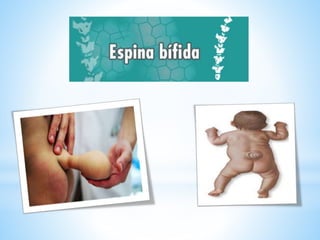
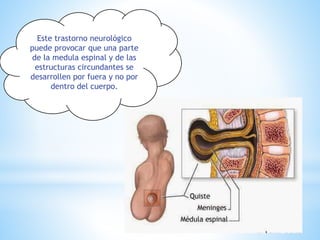
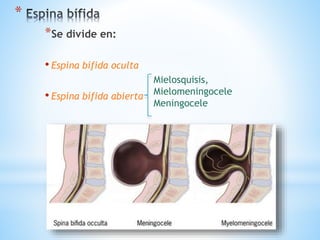
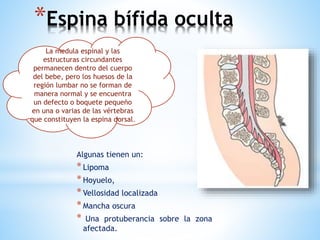
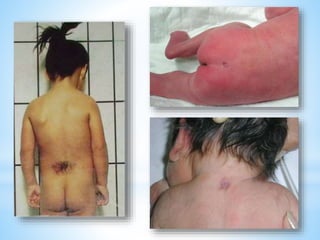
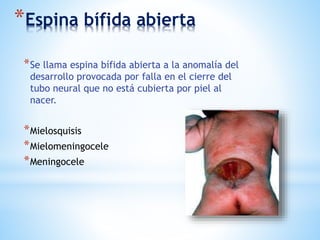
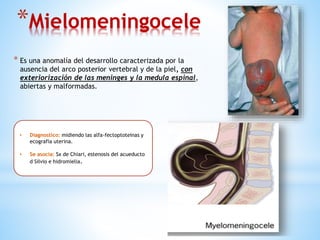
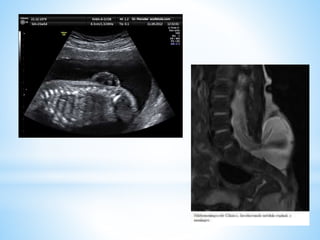
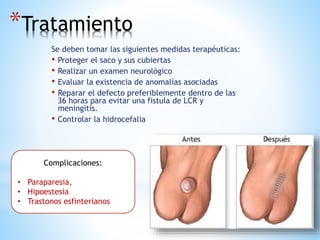
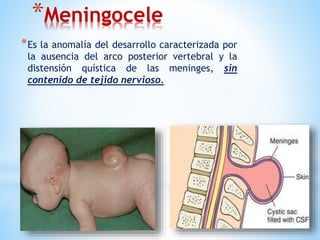
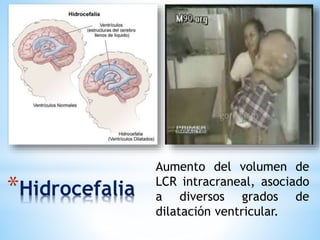
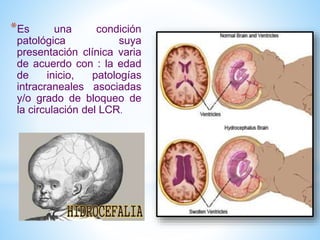
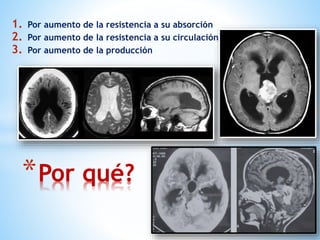
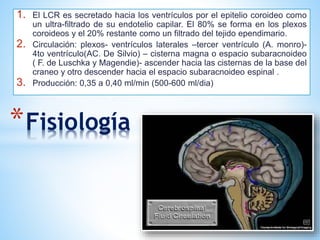
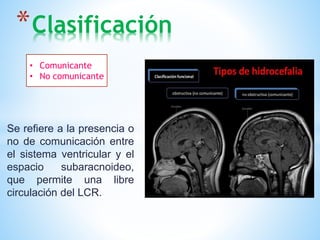
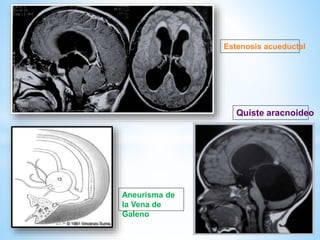
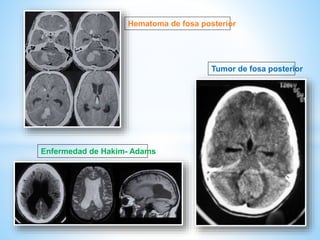
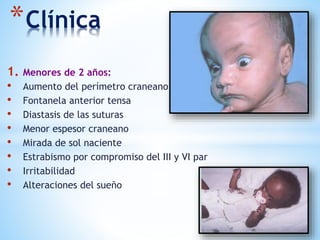
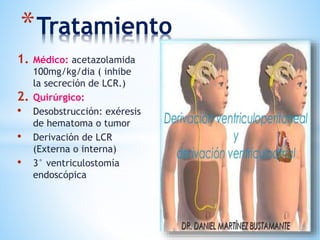
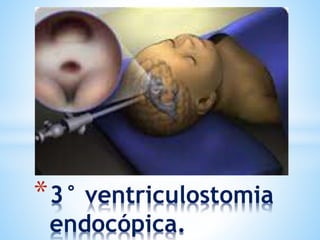
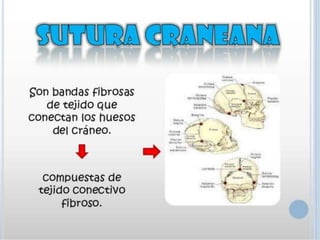
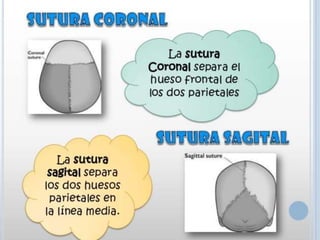
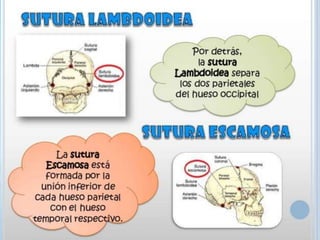
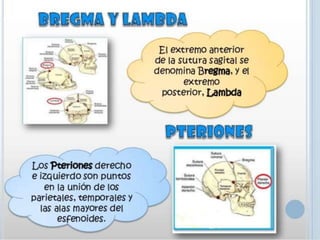
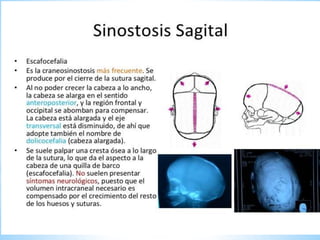
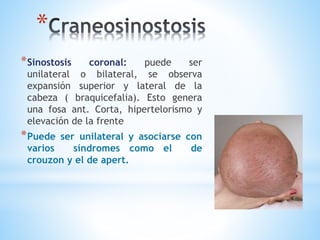
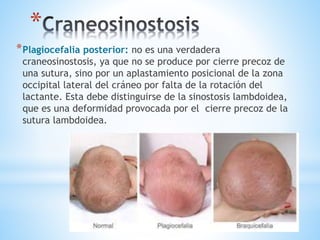
Este documento resume varias malformaciones congénitas del sistema nervioso central. Explica que estas malformaciones ocurren durante el desarrollo embrionario y pueden deberse a factores genéticos, ambientales o infecciosos. Describe defectos como encefalocele, espina bífida y hidrocefalia, explicando sus síntomas, clasificaciones, diagnósticos y tratamientos. También cubre craneosinostosis, resumiendo que implica el cierre prematuro de suturas craneales, lo que afecta el cre