Descargado 223 veces
















![La estenosis hereditaria es transmitida como
un rasgo recesivo ligado a X
Sindrome de Bickers-Adams. [A] Hidrocefalia por estenosis del acueducto
de Silvio; [B] deformidad en flexión y aducción del dedo gordo.
Causas de hidrocefalia congénita](https://image.slidesharecdn.com/malformacionesdelsnc-160330025126/85/Malformaciones-del-snc-19-320.jpg)


















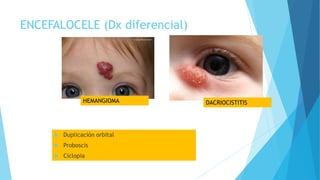




















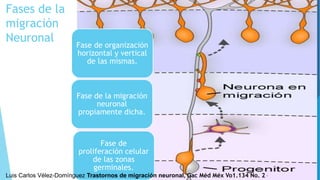


































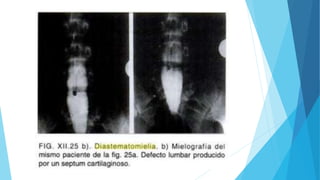

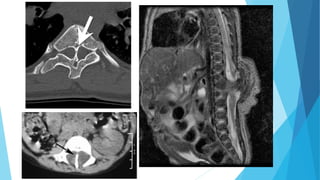








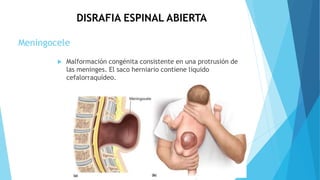





































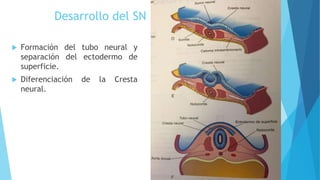
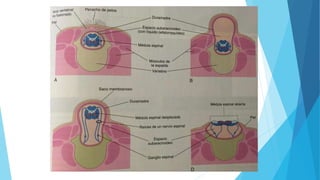
El documento describe varias malformaciones del sistema nervioso central, incluyendo el desarrollo del tubo neural, hidrocefalia, encefalocelia y anencefalia. Explica las etapas del desarrollo del sistema nervioso, desde la formación del tubo neural hasta la diferenciación de las vesículas cerebrales. También describe las causas, síntomas y tratamiento de varias malformaciones como la hidrocefalia y las complicaciones asociadas.













































![Malformaciones_sistema_nervioso_central_[Embriologia]](https://cdn.slidesharecdn.com/ss_thumbnails/malformacionessistemanerviosocentral-100619003148-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)










![Desarrollo fetal[1]](https://cdn.slidesharecdn.com/ss_thumbnails/desarrollofetal1-110304140034-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)


![Desarrollo fetal[1]](https://cdn.slidesharecdn.com/ss_thumbnails/desarrollofetal1-110304140046-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)















































