Descargado 58 veces
























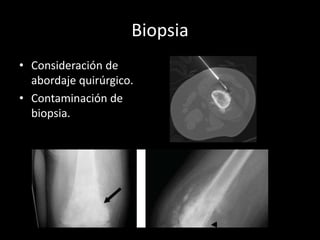



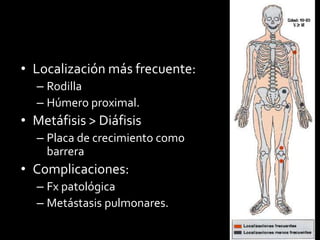





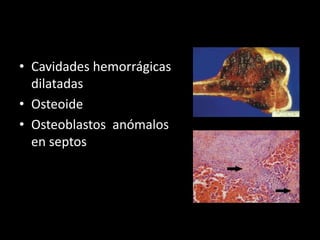




























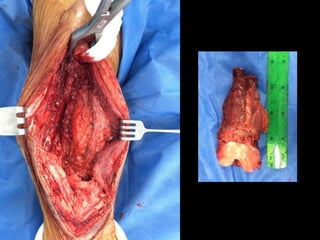








Este documento proporciona información general sobre el osteosarcoma, incluyendo su incidencia, factores de riesgo genéticos, manifestaciones clínicas, pruebas de diagnóstico, clasificación, tratamiento y pronóstico. El osteosarcoma es el sarcoma óseo primario más común que afecta principalmente a adolescentes y adultos jóvenes. Su tratamiento implica quimioterapia neoadyuvante y resección quirúrgica amplia, con buenos resultados si se logra una necrosis tumoral superior al 90%.



































































