
Descargar como PPSX, PPTX









![Fisiopatología↓ síntesis de un tipo de cadena globínicaalfatalasemiarompe el equilibrio Cadenas betabetatalasemia↑ [intracelular] Precipitados intracelulares Cadenas alfadestrucción precoz de los eritroblastos↑Hemolisis eritropoyesis ineficaz↓supervivenciasobrevivenRodakBF. 2005.](https://image.slidesharecdn.com/talasemias-100918233641-phpapp01/85/Talasemias-13-320.jpg)


















![A medida de que reciben transfusiones se sobrecarga de hierro y hemolisis transfucionalToxicdad en órganos parenquimatosos (causa de mayor mortalidad) [Hemosiderosis cardiaca]B talasemia MayorRodakBF. 2005.](https://image.slidesharecdn.com/talasemias-100918233641-phpapp01/85/Talasemias-33-320.jpg)


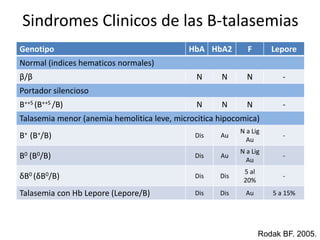



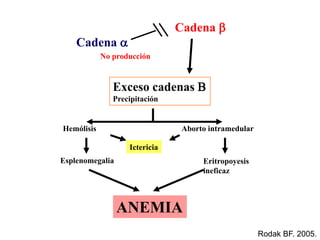











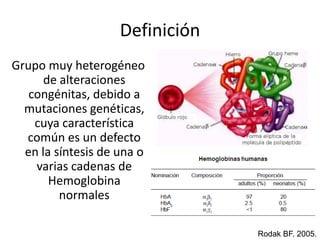
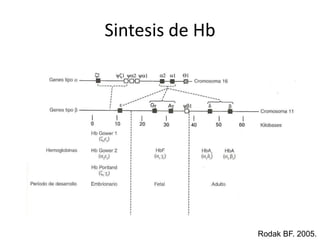
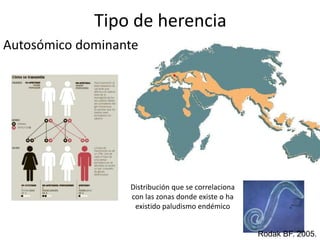
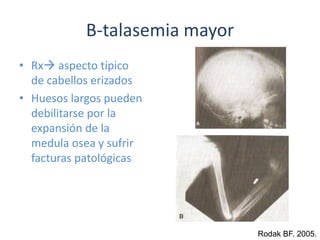
Este documento describe las talasemias, un grupo de trastornos hereditarios caracterizados por un defecto en la síntesis de cadenas de hemoglobina. Se clasifican en beta talasemia y alfa talasemia según la cadena afectada. La beta talasemia incluye trastornos desde portadores asintomáticos hasta talasemia mayor con anemia grave. La alfa talasemia también varía en gravedad desde portadores silenciosos hasta hidropesía fetal fatal. El diagnóstico se basa en estudios hematológicos y genéticos












































































