Descargado 173 veces











![Diagnóstico de amenorrea
secundaria
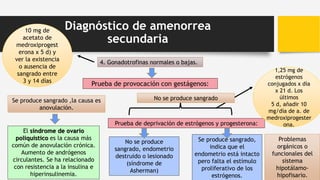
1. Prolactina elevada. Las [ ] altas de prolactina inhiben la secreción pulsátil de
GnRH, lo que conduce a una amenorrea de origen hipofisario.
Se procederá al estudio radiológico de la región selar, mediante TC o RNM.
Los adenomas hipofisiarios secretores de prolactina son la patología
hipofisaria más frecuente que suele cursar con hiperprolactinemia y
amenorrea.
Cerca de la tercera parte de las mujeres con amenorrea secundaria tiene un adenoma
hipofisario.
Otras causas menos frecuentes son el sx de silla turca vacía y otros tumores hipofisarios.
Descartar
hipotiroidismo o
Hiperprolactine
mia yatrogénica.](https://image.slidesharecdn.com/amenorreanuevas-150728160043-lva1-app6892/85/AMENORREA-12-320.jpg)



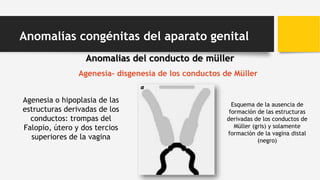



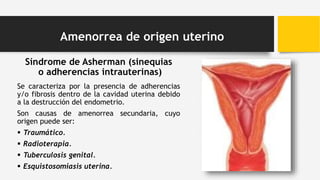








1. La amenorrea se define como la ausencia de menstruación y se divide en primaria y secundaria dependiendo de si ocurre antes o después de la menarquía. 2. Las causas más comunes de amenorrea primaria son el síndrome de Turner, síndrome de Mayer-Rokitansky-Küster-Hauser e insensibilidad a los andrógenos. 3. La causa más frecuente de amenorrea secundaria es el síndrome de ovarios poliquísticos, aunque también pueden deberse a tumores hip











































































































