Descargar para leer sin conexión


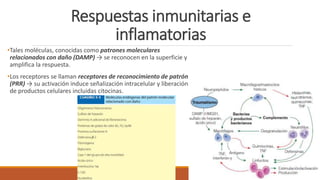








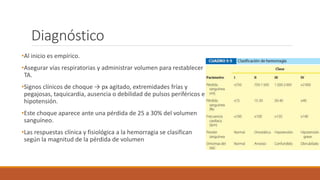





















El documento describe las respuestas inmunitarias e inflamatorias que ocurren durante un choque, incluyendo la liberación de citocinas como TNFα, IL-1, IL-6 e IL-10. También describe las formas de choque como hipovolémico, séptico y cardiógeno, así como sus causas, diagnósticos y tratamientos.



































































