Descargado 345 veces






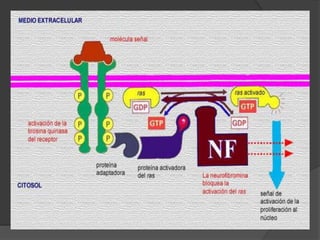
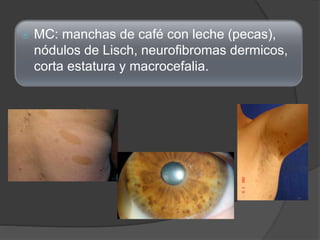




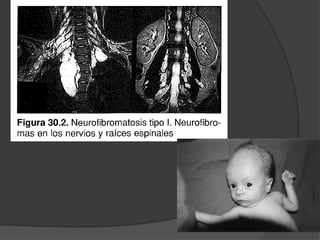



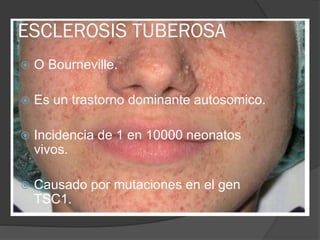








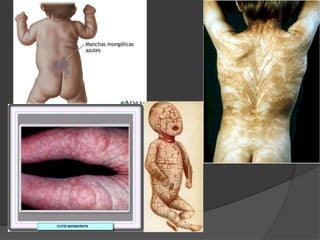

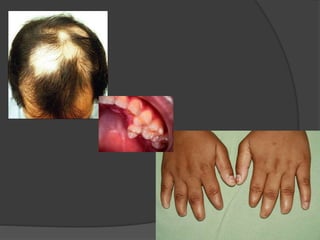
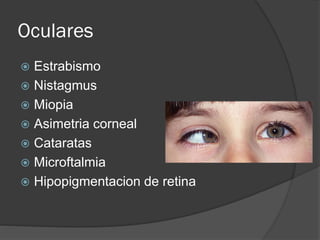






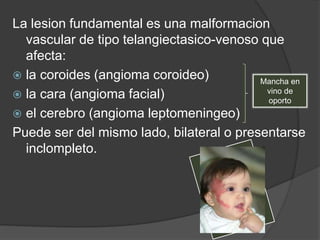
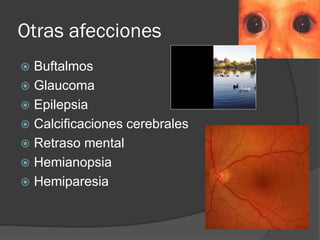

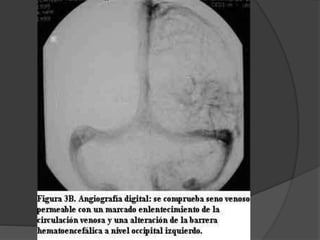
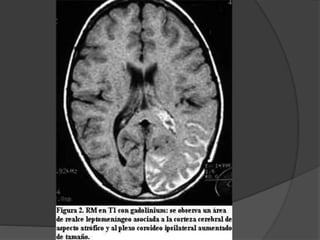
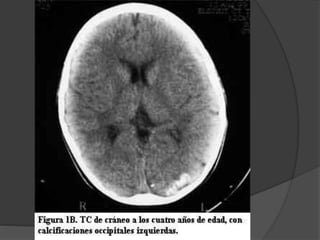




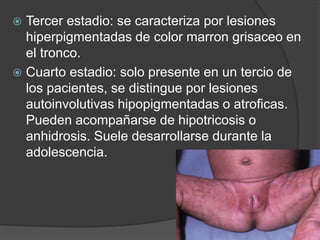
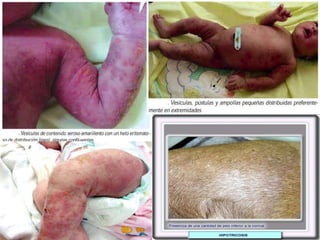




El documento detalla diversas facomatosis, que son trastornos hereditarios caracterizados por anomalías del desarrollo embrionario y la aparición de manchas y tumores, destacando condiciones como la neurofibromatosis y la esclerosis tuberosa. Se describen sus causas, incidencias, manifestaciones clínicas y complicaciones, además de abordar el diagnóstico y tratamiento de cada una. También se mencionan otras condiciones neurocutáneas como la hipomelanosis de Ito y el síndrome de Sturge-Weber, junto con su etiología, síntomas y opciones terapeuticas.










































































