Descargado 89 veces



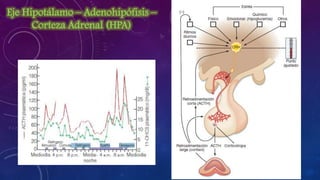
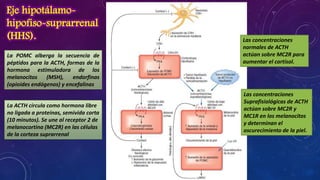
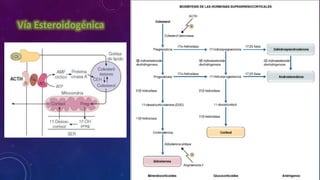
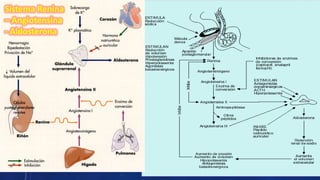
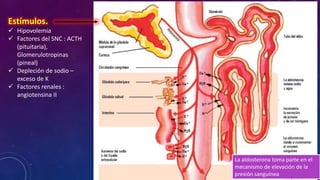





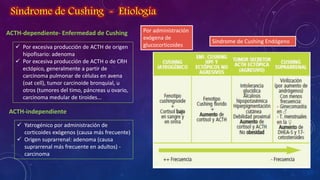
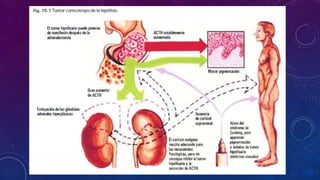
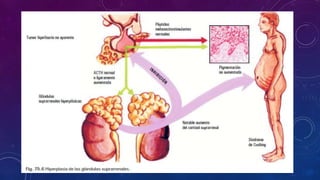
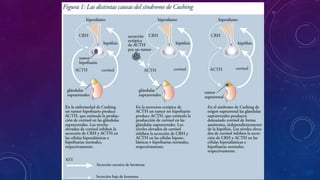













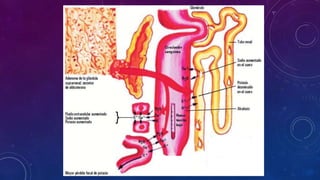



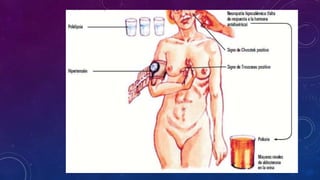




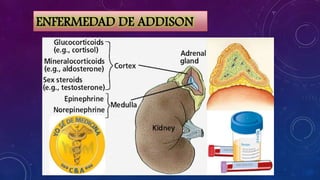




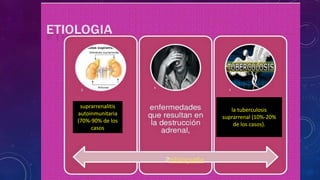







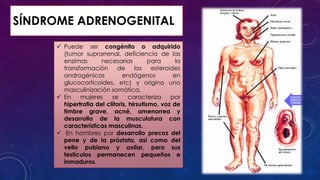





El documento resume la fisiopatología de la corteza suprarrenal. La corteza suprarrenal está dividida en tres zonas que secretan diferentes hormonas. La zona glomerular secreta mineralocorticoides como la aldosterona, la zona fascicular secreta glucocorticoides como el cortisol, y la zona reticular no tiene función endocrina conocida. Los principales síndromes asociados con alteraciones en la corteza suprarrenal son el síndrome de Cushing debido a exceso de glucocorticoides, y el hiperaldosteronismo











































































































