Descargado 97 veces
















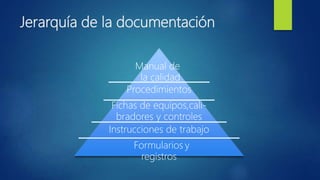
































El documento describe los requisitos para la acreditación de laboratorios médicos según la norma ISO 15189. Establece que la norma especifica los requisitos de calidad y competencia para laboratorios clínicos, incluyendo un sistema de gestión de calidad documentado, requisitos para el personal, las instalaciones, los equipos, y los procedimientos pre-analíticos y analíticos. El objetivo es promover un enfoque común para la gestión de la calidad en laboratorios médicos y mejorar los servicios para los pacientes.






























































































