Descargado 766 veces


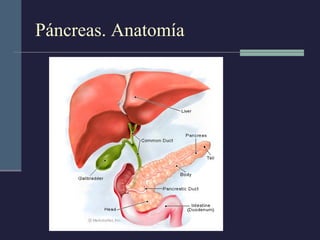
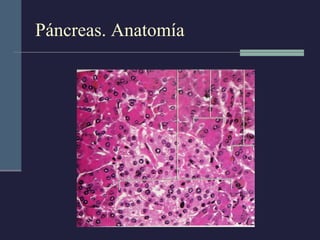
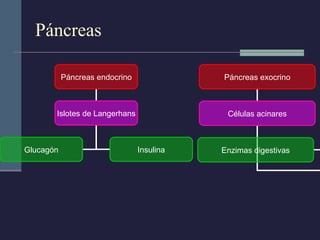
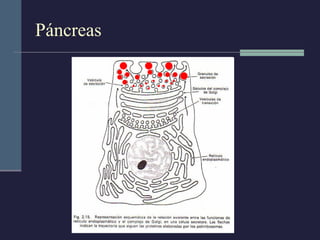
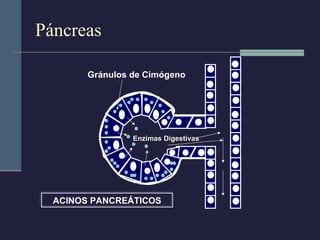

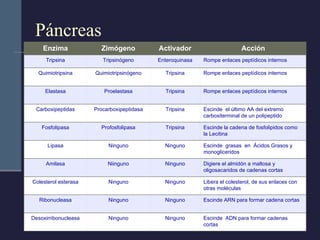

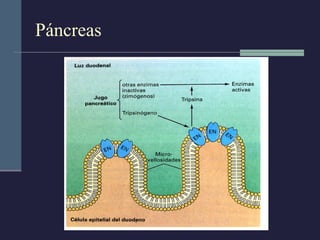
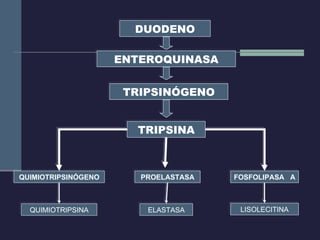


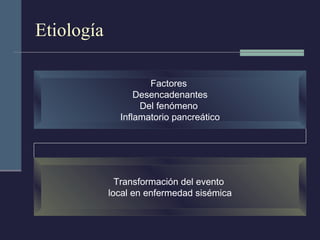
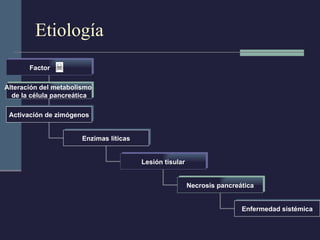

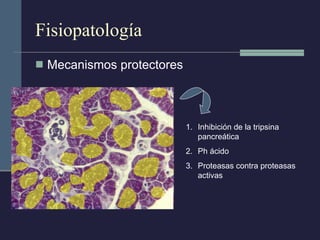

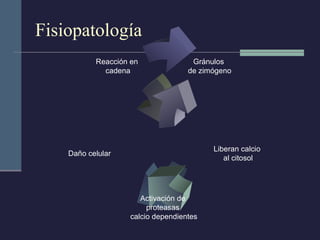
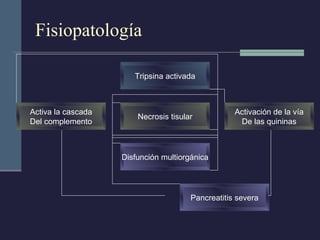


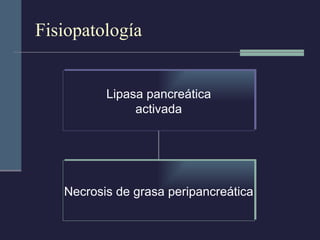
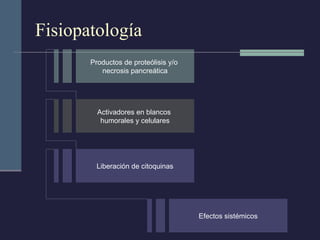
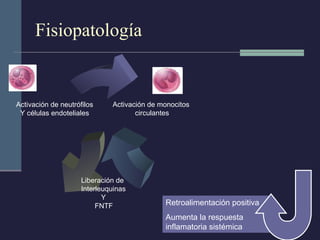
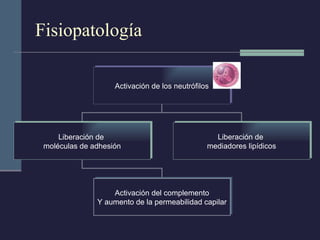
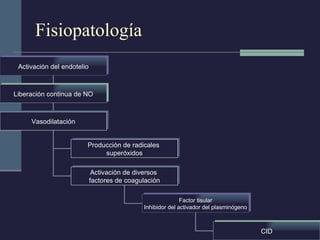
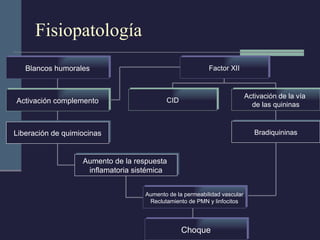
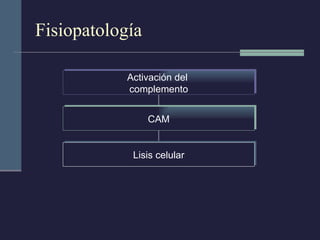
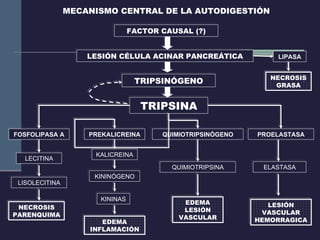

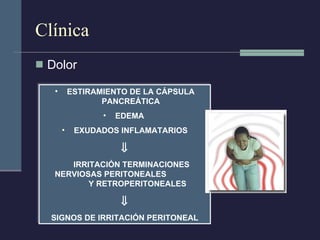
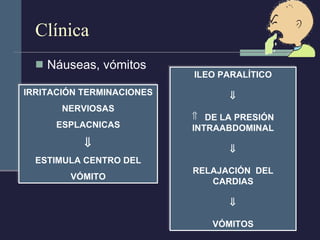

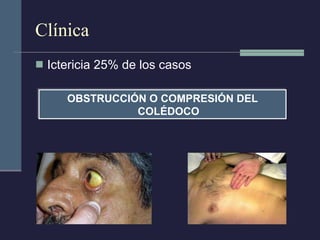
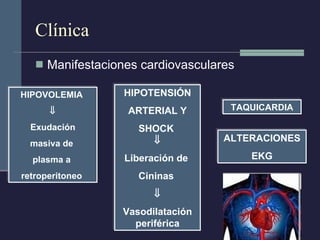
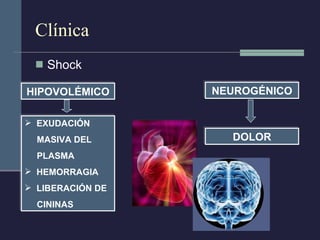








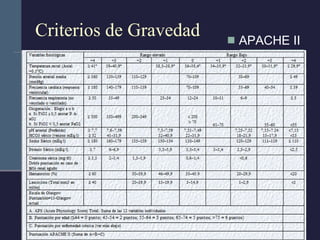
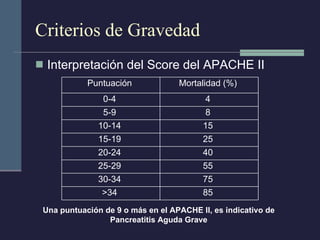










Este documento resume los conceptos clave de la pancreatitis, incluyendo su definición, etiología, mecanismos fisiopatológicos, manifestaciones clínicas, criterios de gravedad y pronóstico. Describe la anatomía y función del páncreas, así como los mecanismos por los cuales la activación prematura de enzimas digestivas conduce a daño pancreático y sistémico en la pancreatitis aguda. También explica las teorías sobre la progresión a pancreatitis crónica debido a episodios repetidos o factores

































































































