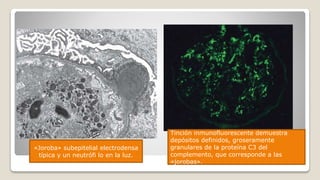
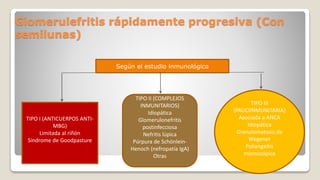
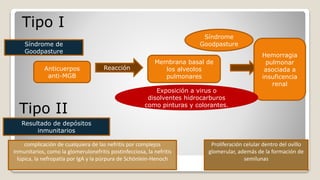
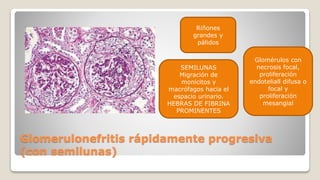
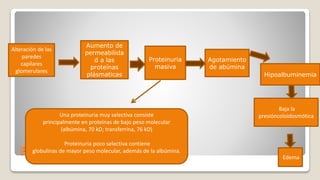
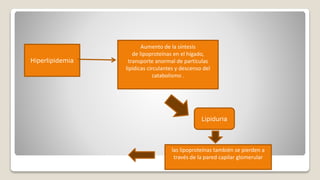
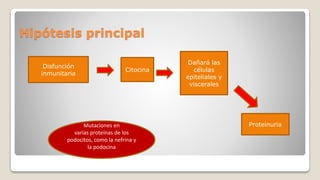
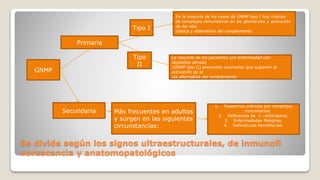
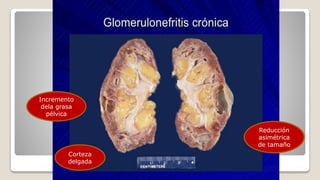
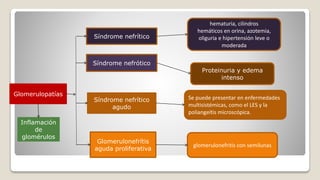
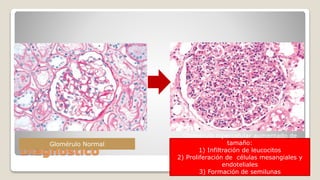
El documento revisa el síndrome nefrítico, describiendo su concepto, mecanismos patogénicos y clasificación, así como las diversas formas de glomerulonefritis. Se destacan los procesos de formación de orina en los riñones y las características del glomérulo y otros componentes renales. Además, se abordan las diferencias entre el síndrome nefrítico y el síndrome nefrótico, incluyendo la fisiopatología y las complicaciones asociadas.





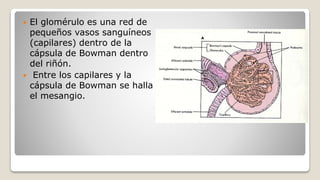
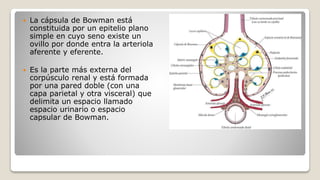





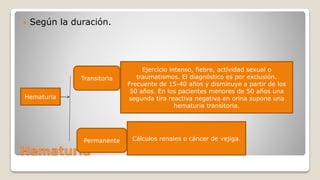
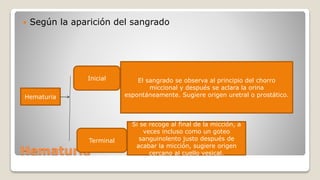



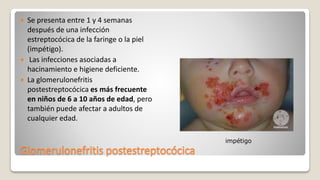

![Glomerulonefritis aguda no estreptocócica
(glomerulonefritis postinfecciosa)
En relación con otras infecciones, bacterianas:
endocarditis estafilocócica, neumonía neumocócica y meningococcemia.
Víricas:
Hepatitis B, hepatitis C, parotiditis, infección por el virus de la
inmunodeficiencia humana [VIH], varicela y mononucleosis infecciosa.
Parasitarias: malaria, toxoplasmosis.
Esporádica](https://image.slidesharecdn.com/patologaequipo5glomerulonefrtis11-170620175542/85/Patologia-glomerulonefritis-25-320.jpg)