
Descargado 40 veces










































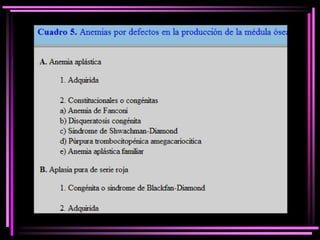



















El documento proporciona información sobre las anemias. Define la anemia y explica sus mecanismos fisiopatológicos. Clasifica las anemias de acuerdo a su velocidad de instalación, patogenia y morfología. Describe en detalle la anemia ferropénica, la anemia megaloblástica y las anemias por defectos en la producción de la médula ósea.






































![Trabajo De Vir Y Cynthia[1] Terminado[1]](https://cdn.slidesharecdn.com/ss_thumbnails/trabajodevirycynthia1terminado1-090525030127-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)




































































