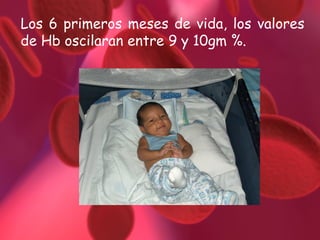
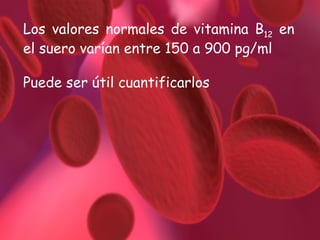
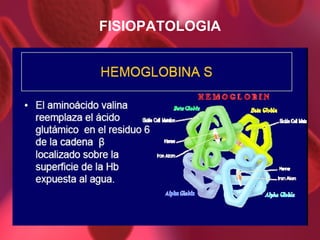
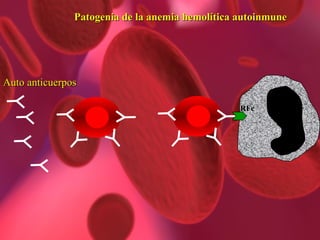
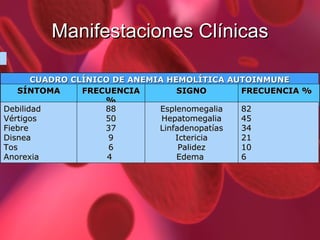
El documento proporciona información sobre diferentes tipos de anemia. Explica que la anemia ferropénica es la causa más común de anemia en lactantes y preescolares, debido a una dieta baja en hierro. También describe la anemia megaloblástica, causada por deficiencias de vitamina B12 o ácido fólico, y su papel en la síntesis de ADN. Además, explica la fisiopatología y los tipos de anemia falciforme, un trastorno hereditario que afecta la forma de los gl