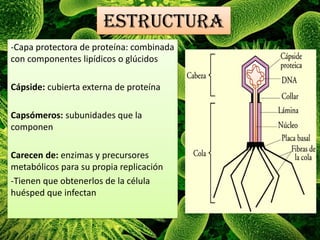
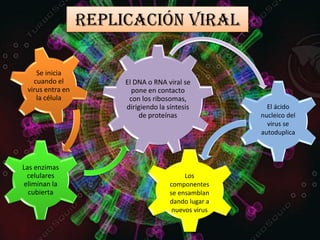
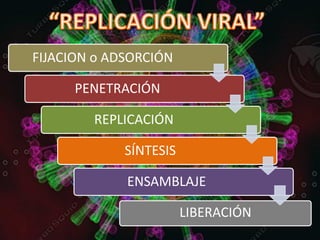
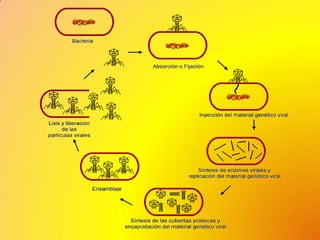
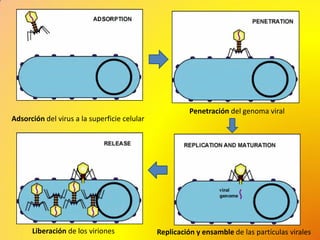
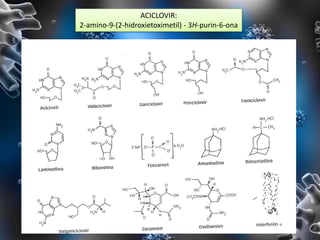
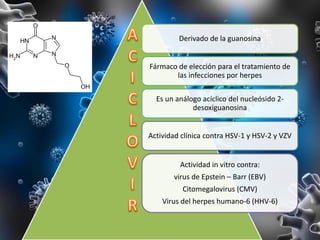
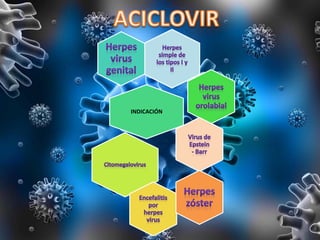
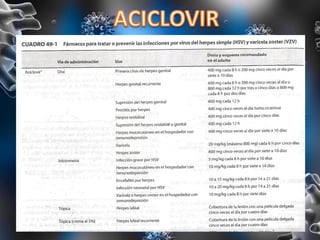
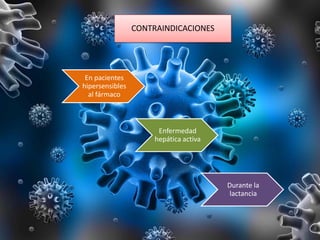
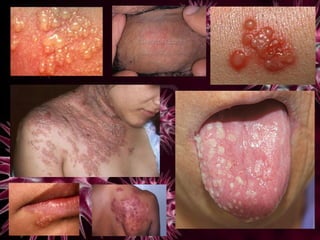
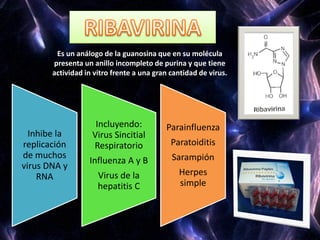
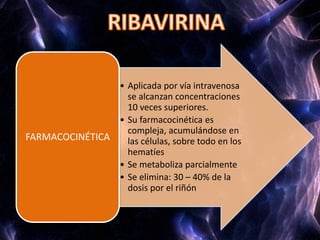
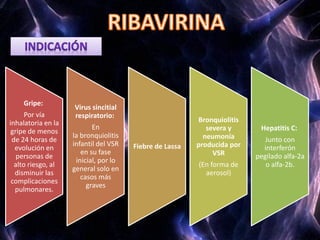
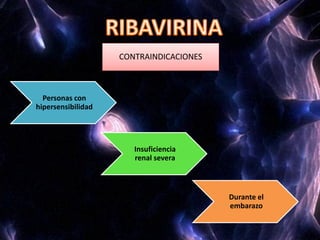
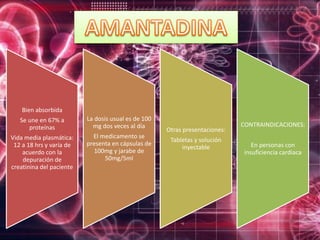
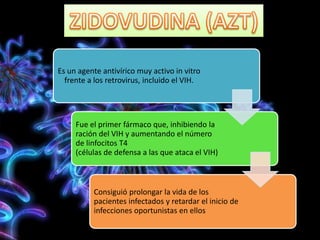
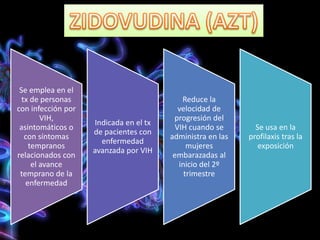
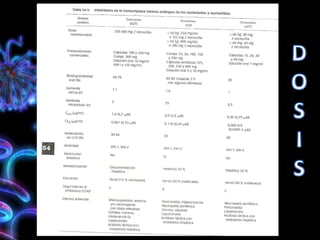
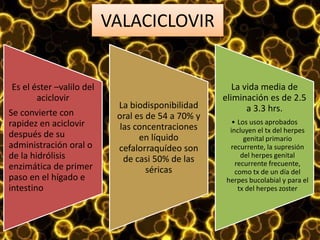
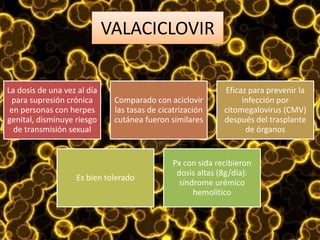
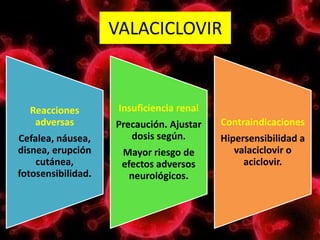
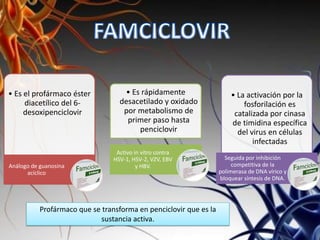
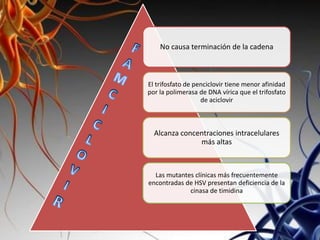
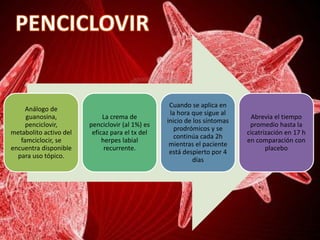
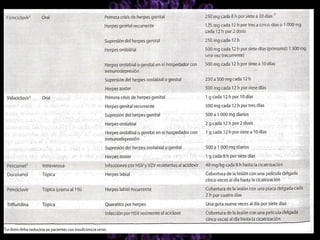
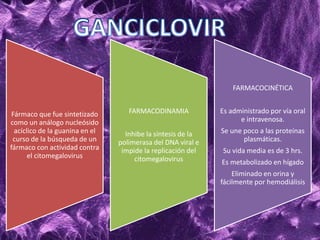
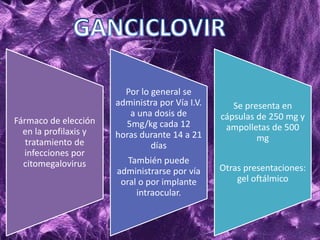
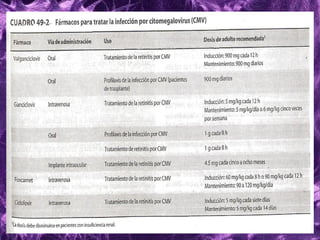
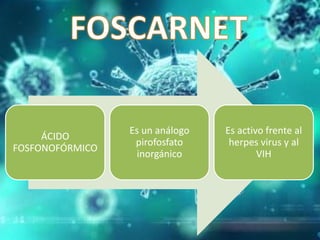
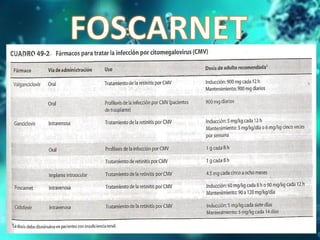
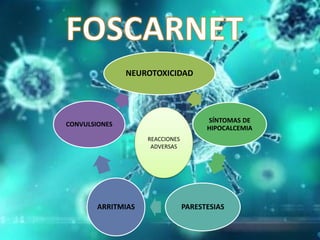
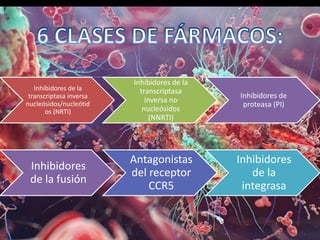
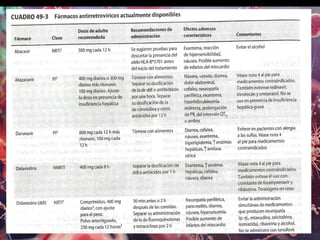
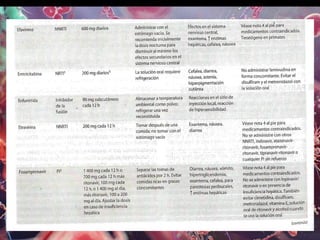
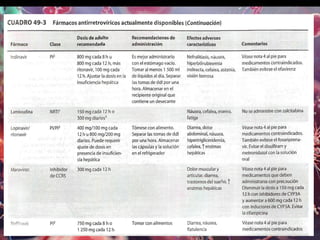
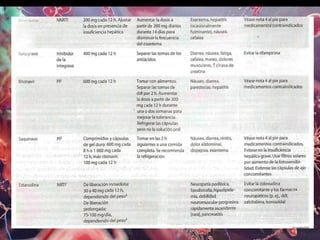
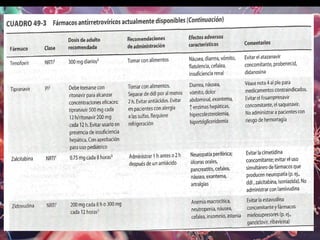
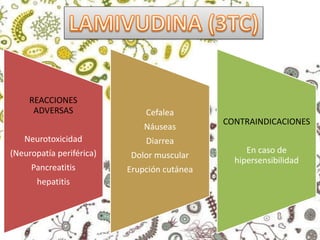
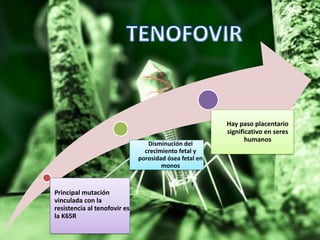
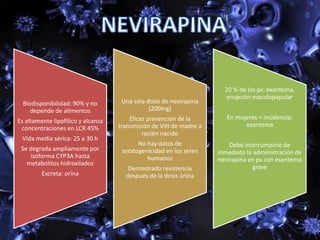
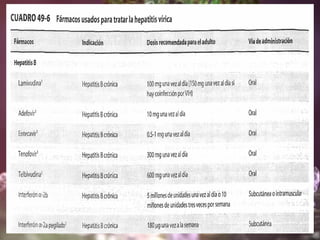
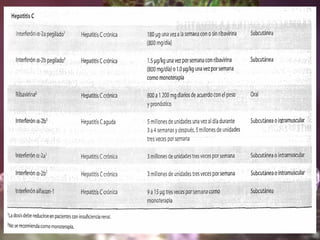
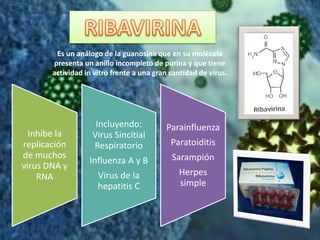
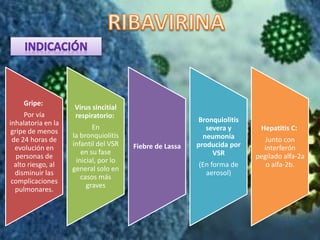
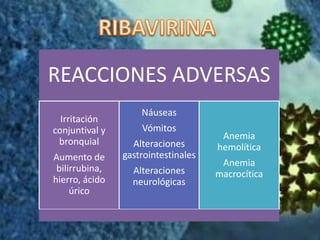
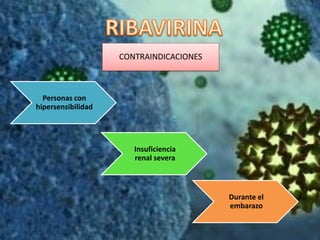
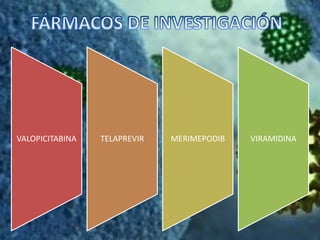
Los virus son agentes infecciosos obligados que sólo se replican dentro de células vivas, careciendo de metabolismo propio y compuestos necesarios para su replicación. Los antivirales son medicamentos que tratan infecciones virales, actuando en varias etapas de la replicación del virus y algunos de ellos requieren activación en el interior de la célula infectada. El aciclovir, un antiviral importante, se usa principalmente contra infecciones por herpes y actúa inhibiendo la polimerasa de DNA viral, mientras que otros antivirales, como la ribavirina y la amantadina, tienen mecanismos de acción diferentes.