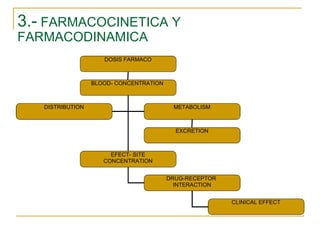
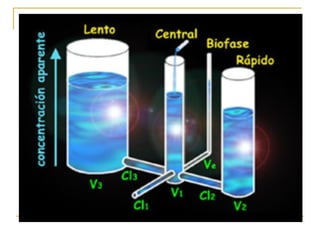
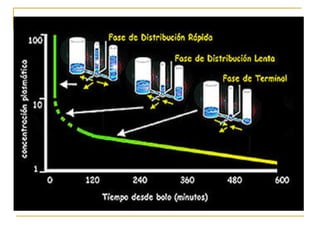
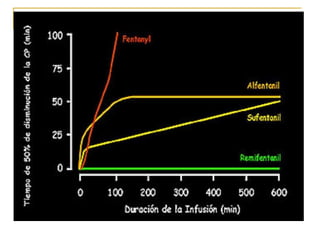
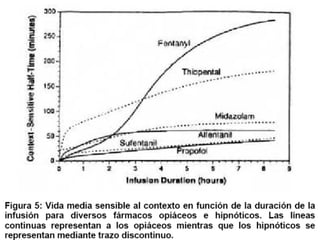
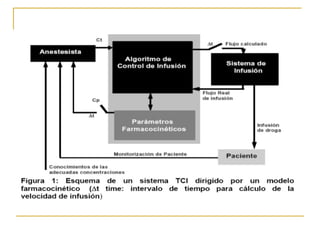
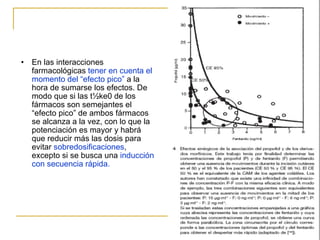
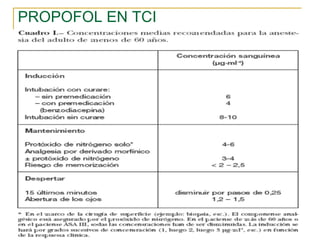
Este documento resume los principios generales de la anestesia intravenosa. Explica que la mayoría de los fármacos anestésicos siguen un modelo farmacocinético tricompartimental donde se distribuyen en tres compartimentos: central, periférico rápido y periférico lento. También describe conceptos como volumen de distribución, aclaramiento y tiempo de vida media, así como las ventajas e inconvenientes de la técnica de anestesia total intravenosa.





































































































![02.7 Toxicocin%C3%A9tica 1[1]](https://cdn.slidesharecdn.com/ss_thumbnails/02-7toxicocinc3a9tica11-100114212335-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



























































