Descargado 208 veces



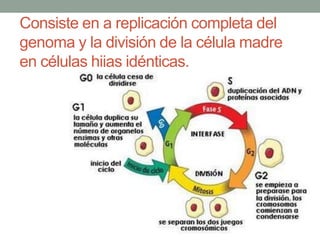
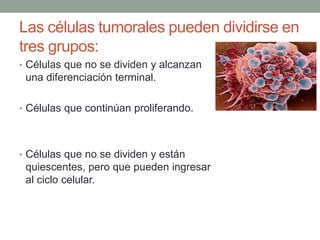



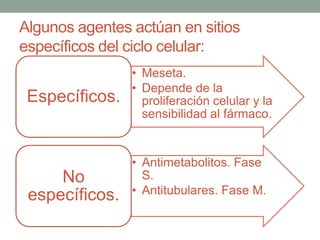














La quimioterapia consiste en el uso de fármacos para tratar el cáncer. Existen varios tipos de agentes quimioterapéuticos que actúan en diferentes puntos del ciclo celular como las fases S y M. La quimioterapia puede ser específica para ciertas fases o no específica, y se utilizan esquemas multiagentes para maximizar la eficacia del tratamiento. La determinación de la respuesta al tratamiento se realiza mediante criterios como RECIST.














































































