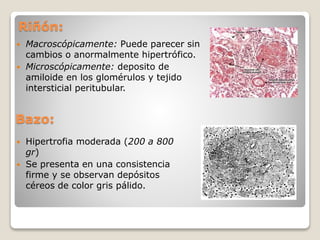
La amiloidosis es una afección causada por depósitos extracelulares de proteínas fibrilares anormalmente plegadas que dañan los tejidos. Puede ser sistémica cuando afecta múltiples órganos, o localizada cuando se limita a un solo órgano. Las proteínas amiloides más comunes son la cadena ligera de amiloide AL, la fibrilla AA y la proteína beta amiloide. La clasificación dependiendo de la causa incluye amiloidosis primaria asociada a mieloma múltiple




































































![Expo Pato Testiculo[1]](https://cdn.slidesharecdn.com/ss_thumbnails/expopatotesticulo1-091106094855-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)









![Vacuna contra la rabia humana[1]](https://cdn.slidesharecdn.com/ss_thumbnails/vacunacontralarabiahumana1-100824085733-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




