Descargado 136 veces


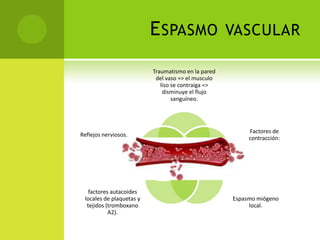
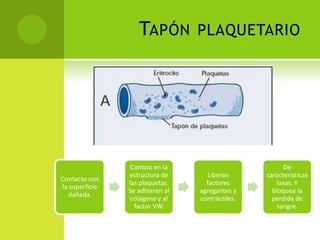



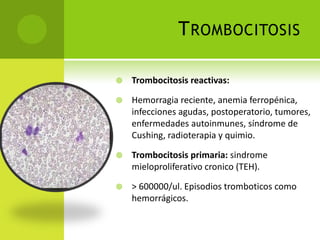









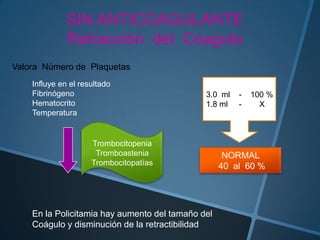































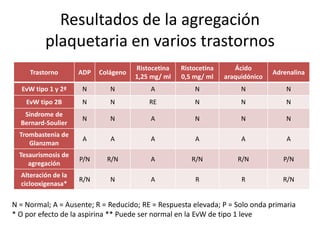








Este documento proporciona información sobre varias pruebas de coagulación sanguínea como el tiempo de tromboplastina, pruebas de función plaquetaria y tiempo de sangrado. Describe los procedimientos, valores normales e interpretación de los resultados de estas pruebas que evalúan la hemostasia y la función plaquetaria.











































































































