Descargado 304 veces









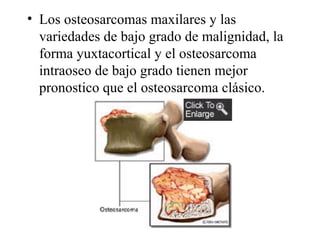








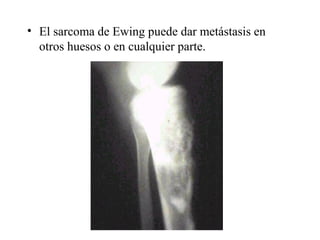






Este documento describe varios tipos de tumores óseos malignos. Los tumores óseos malignos primarios son poco frecuentes y más comunes en hombres jóvenes. Los tumores óseos malignos más comunes son osteosarcomas, condrosarcomas, fibrosarcomas y sarcomas de Ewing. Estos tumores se originan en áreas de crecimiento óseo rápido y pueden causar dolor, hinchazón y metástasis, especialmente en los pulmones. El tratamiento incluye cirugía, radioterapia y























































































