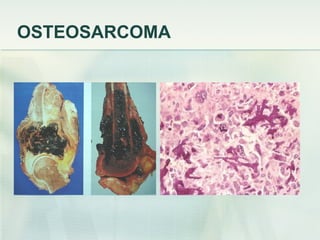
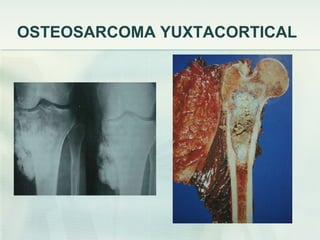
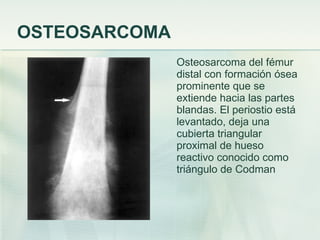
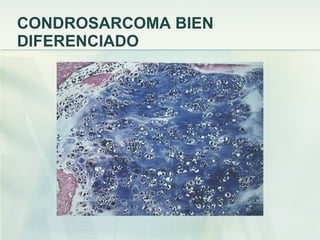
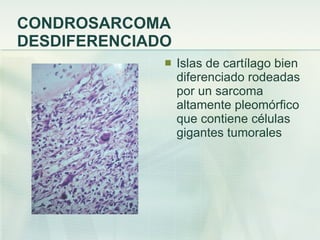
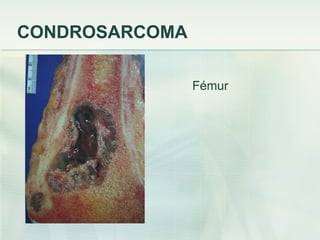
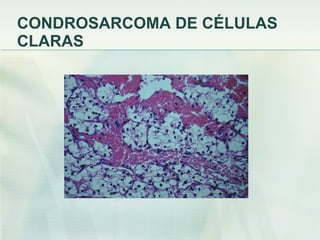
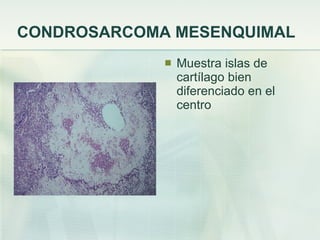
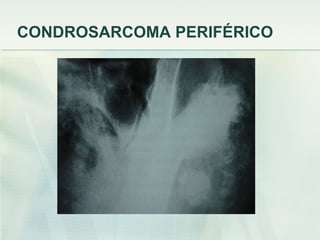
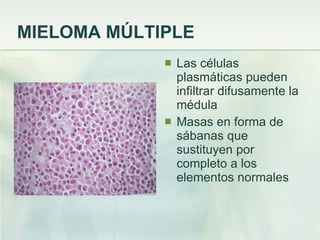
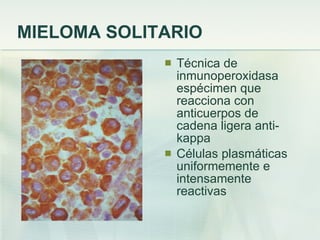
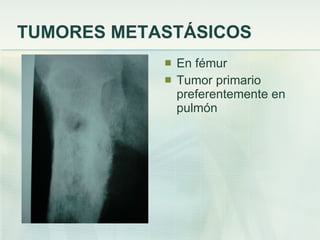
El documento describe los tumores óseos malignos más comunes, incluyendo el osteosarcoma, condrosarcoma y sarcoma de Ewing. El osteosarcoma es el tumor óseo maligno primario más frecuente, que se origina en las metáfisis de los huesos largos. El condrosarcoma es el segundo tumor productor de matriz ósea más común, que produce cartílago neoplásico. El sarcoma de Ewing afecta principalmente a niños y jóvenes, y se caracteriza por translocaciones cromosómicas específicas