Descargado 319 veces


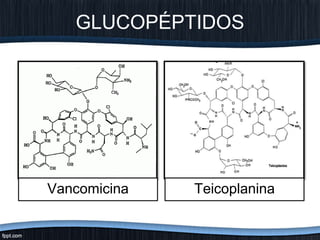



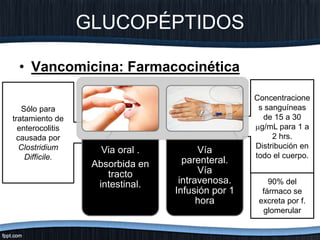




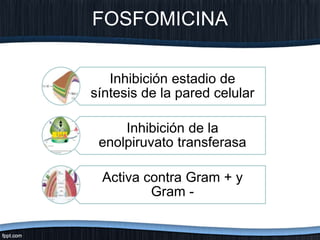



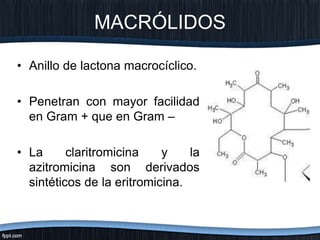



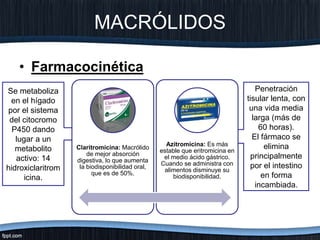


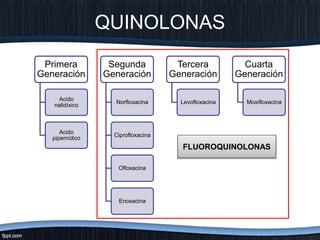

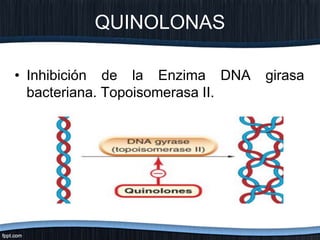
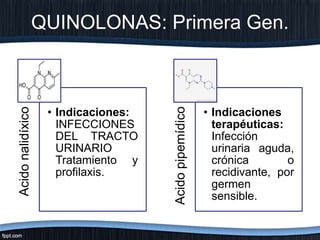

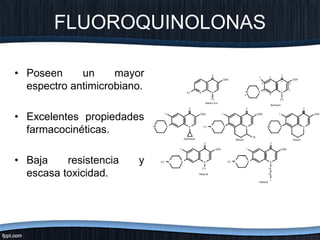

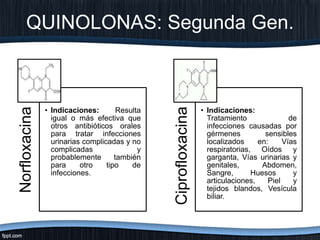
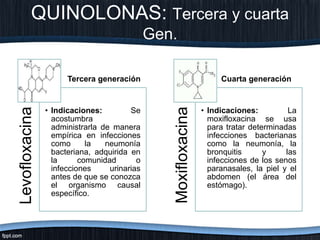



El documento trata sobre diferentes tipos de antibióticos glucopeptidos y cómo actúan. Explica que la vancomicina es el antibiótico glucopeptídico clásico que actúa uniéndose a la pared bacteriana e inhibiendo su formación, mientras que la teicoplanina es más reciente y mejora algunos aspectos de la vancomicina. También resume brevemente las características de estos medicamentos.




























































































