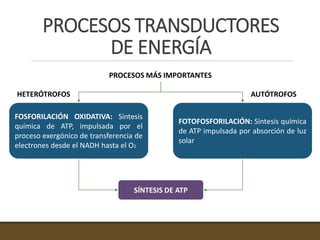
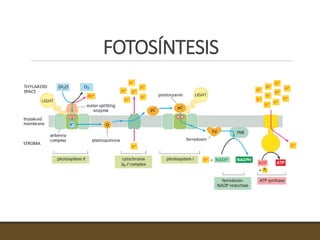
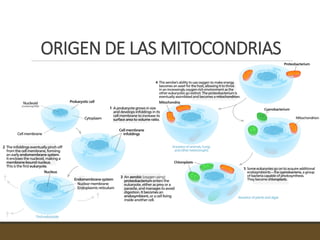
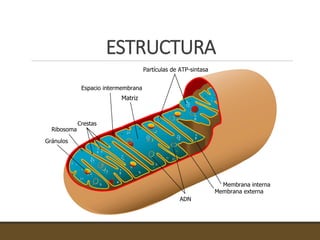
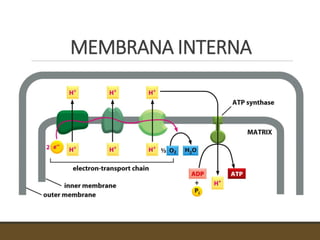
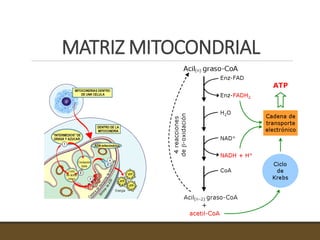
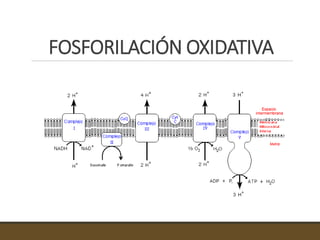
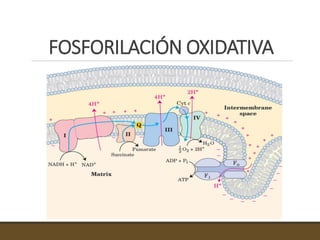
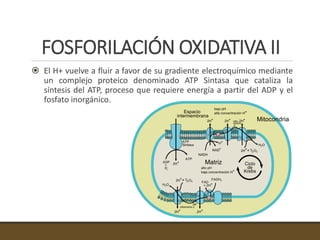
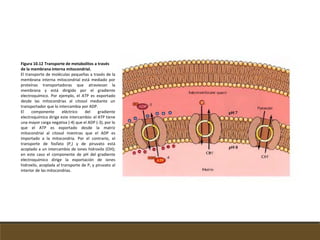
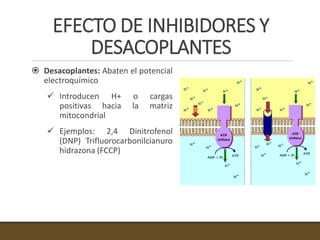
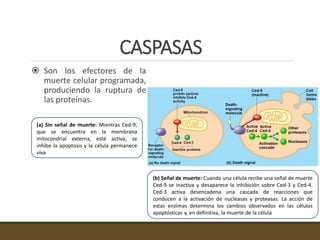
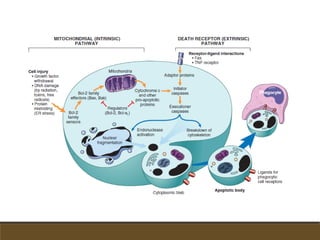
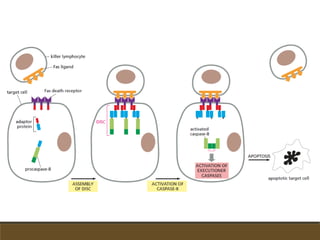
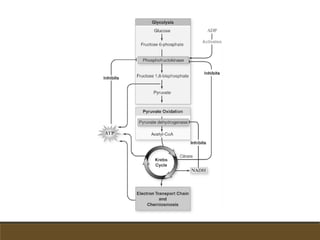
El documento trata sobre la transducción de energía en las mitocondrias y cloroplastos. Brevemente describe que: 1) Las mitocondrias y cloroplastos evolucionaron de bacterias fagocitadas por células eucariotas primitivas. 2) Utilizan procesos quimio-osmóticos para producir ATP acoplando la transferencia de electrones a un bombeo de protones. 3) En las mitocondrias, la fosforilación oxidativa genera un gradiente de protones a través de la membrana interna que se usa