Descargado 5662 veces
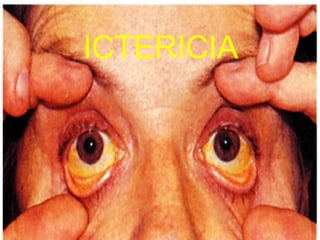



















































































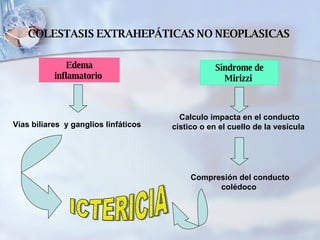
























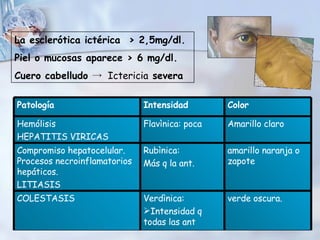



















El documento describe la anatomía y fisiología del hígado, así como las causas y clasificación de la ictericia. Explica que la ictericia se produce por el aumento de bilirrubina en la sangre, y clasifica las causas en hepáticas, obstructivas y hemolíticas. Detalla los procesos de metabolismo de la bilirrubina, transporte y excreción a través del hígado.





































































































