Descargado 172 veces







































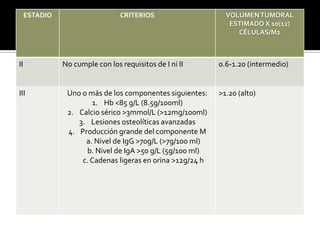





































Este documento describe el caso clínico de una paciente femenina que presenta dolor óseo, anemia y lesiones óseas líticas. Se realizaron varios exámenes que mostraron elevación de gammaglobulinas, presencia de cadenas ligeras en orina, y hallazgos en la médula ósea consistentes con mieloma múltiple. El diagnóstico final fue mieloma múltiple basado en los criterios mayores y menores establecidos.

















































































