Sx mieloproliferativos
•Descargar como PPTX, PDF•
1 recomendación•1,112 vistas

medicina

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Destacado (20)
Similar a Sx mieloproliferativos
Similar a Sx mieloproliferativos (20)
Más de Jorge Ezequiel Hernandez Partida
Más de Jorge Ezequiel Hernandez Partida (20)
Último
Último (20)
Sx mieloproliferativos
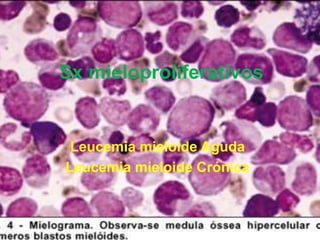
- 1. Sx mieloproliferativos Leucemia mieloide Aguda Leucemia mieloide Crónica
- 2. Leucemia Mieloide Aguda • Historia: • En 1827, cuando Alfred-Armand Marie Velpeau describió el caso de una florista de 63 años de edad con una enfermedad cuyos principales síntomas eran fiebre, debilidad, cálculos renales y hepatosplenomegalia
- 3. • En 1845, el patólogo J.H. Bennett reportó una serie de casos similares de pacientes que fallecieron con esplenomegalia y cambios "en el color y la consistencia de la sangre". • Bennett utilizó el término "leucocitemia" para describir esta condición patológica. • Virchow fue el primero en describir el anormal exceso de glóbulos blancos en pacientes con el síndrome clínico descrito por Velpeau y Bennett.
- 4. • En 1877, Paul Ehrlich desarrolló técnicas de tinción de células sanguíneas que le permitieron describir en detalle y diferenciar los glóbulos blancos normales y anormales. • Wilhelm Ebstein introdujo el término "leucemia aguda"
- 5. • El término "mieloide" fue acuñado por Neumann en 1869, tras ser el primero en determinar que los glóbulos blancos provenían de la médula ósea (µυєλός = médula). • Finalmente, Naegli caracterizó los mieloblastos, que pertenecen a la estirpe celular afectada en la LMA, y dividió los tipos de leucemia en mieloides y linfoides.
- 8. • Estas enfermedades afectan a la medula y en menor grado a los órganos hematopoyéticos secundarios. • Hay alteración en la hematopoyesis • La hematopoyesis va siendo regulada por citocinas, factores de crecimiento y cascadas de activación enzimática, una alteración será el causante de la patología.
- 9. Categorías de neoplasias mieloides • Leucemias mieloides (donde hay acumulación de blastos en medula) • Sx Mielodisplasicos (la hematopoyesis ineficaz provoca citopenias) • Transtornos mieloproliferativos (hay aumento en la producción hematopoyética).
- 10. • La leucemia es un tumor de progenitores hematopoyéticos (cels. pluripotenciales) que impiden la diferenciación hematopoyética (anemia, trombocitopenia) y sobreproducción de cels inmaduras • Provocan la acumulación de blastos en la medula. • La leucemia se presenta mas a partir de los 60 años (13 000 casos al año)
- 11. Clasificación de LMA por FAB M0 Leucemia con mínima diferenciación 5% M1 Mínima inmadura 20% M2 Mieloblastica madura 30% M3 Promielocitica granular 10% M4 Mielomonocitica 20% M5 Monocitica pura 10% M6 eritroleucemia 4% M7 megacarioblastica 1%
- 15. Leucemia Linfoide Aguda • L1 (linfoblastica típica)= cels. blasticas pequeñas, núcleo redondeado sin nucléolo y con escaso citosol. • L2 (linfoblastica atípica)= cels. Blasticas grandes, núcleo irregular y abundante citosol • L3 = cels. Blasticas grandes, núcleo redondeado con nucléolo prominente y vacuolas
- 16. Morfología de LMA • El Dx se basa en la presencia de 20% de blastos mieloides en medula (pueden ser >100,000, <10,000 o estar ausentes) • Los bastones de Auer (inclusiones azurofilas) son patognomónicos de la LMA. • CD33 (inmadurez) CD15 (madurez)
- 19. Descripción morfológica • 1-mieloblastos= cuentan con 2 a 4 nucléolos y gránulos azurofilos (peroxidasa +) y bastones de Auer. • 2-monoblastos= nucléolos plegados, estereasa + • También suele haber megacarioblastos.
- 22. Casusas de LMA • La causa precisa del porque la leucemia se desconoce (se piensa que es multifactorial): • Se piensa que se debe a: • Herencia cromosómica • Exposición a Radiaciones • Exposición a sustancias químicas • Quimioterapéuticos.
- 23. Herencia • Mayor incidencia de aparición de LMA en Síndromes como: • Ataxia Telangiectasia, Sx Down, Anemia de Fanconi • Sx de Bloom (enanismo, rostro afilado y abundantes telangiectasias cutáneas). • Genéticamente se observa una extremada fragilidad de los cromosomas.
- 24. • Suele haber mutaciones de P53, translocaciones y genes quiméricos. • Se observo mayor incidencia de leucemia en la población japoneses que sobrevivió a Hiroshima y Nagasaki • Al paso de años los antineoplásicos tienden a generar leucemia (presentan deleciones en cromosomas 5 y 7) (ciclofosfamida, antraciclinas)
- 25. • Sustancias químicas( benceno y plaguicidas como DDT y Paraquat y nicotina) • Hay 2 anomalías cromosómicas bien establecidas: • 1- t(15;17) (q22;q12) con Leucemia Aguda Promielocitica (M3)
- 26. • 2- inv(16) (p13q22) con LMA de eosinofilos anormales. • Y la leucemia M7 con CD41 y CD61 • Se alteran genes que codifican los factores de transcripción necesarios para la diferenciación mieloide.
- 27. • Los dos reordenamientos mas comunes son t(8;21) e inv(16) codifican proteínas de fusión que interfieren con la función de CBF1a/CBF1b(factor de transcripción necesario para la hematopoyesis). • t(8;21)--------altera al gen CBF1a y inv(16)—lo hace con CBF1b • Ambos genes necesarios para codificar el CBF1a/CBF1b obligado para la maduración.
- 28. • Y también se implica la anormal actividad de las tirosinsinasas (c-Kit) • Otra alteración es la de: • t(15;17) produce a PML que se une a RARa (receptor a del ac. Retinoico) formando la proteína de fusión PML/RARa • Esta interacciona con represores transcripcionales que suprimen la diferenciación celular
- 29. t(15;17) también produce mutación en el receptor de tirosincinasa FLT-3 (diferenciación) • Las leucemias con la translocacion t(15;17) responden muy bien al Acido Alltransretinoico (ATRA) (se une a PML- RARa)
- 30. Manifestaciones clínicas • 1- hay cansancio, perdida de peso y hemorragias, hematomas e infecciones frecuentemente y fiebre • Dolor óseo, cefalea, adenopatía, tos y transtornos visuales y cefalea. • Puede presentarse como mieloblastoma, cloroma o sarcoma granulocitico
- 31. • También hay infiltrado blastico a otros tejidos como las encías, piel y retina entre otros. • Hepatosplenomegalia es característica de LMA y la leucocitosis • BCR-ABL-Leucemia Linfoblastica • PML-RARa-Leucemia Promielocitica Aguda (M3) TX-Ac. Alltransretinoico • AML-ETO-Leucemia mieloblastica Aguda (M1, M2)
- 32. Leucemia Monocitica (M5) en piel
- 33. LMA en Laboratorio • 1-presencia de cels. Blasticas > 20% de la celularidad total • 2-anemia normocitica normocromica y trombocitopenia < 100 000/mm3 • 3-leucocitosis >15 000/mm3 (o normal o leucopenia) • 4-VSH acelerada y colesterol bajo • 5-aumeto moderado de Ac. Úrico
- 34. DX de LMA • 1. estudio de sangre o aspirado de medula ósea con tinción Giemsa, May Gronwald y Romanowski. • 2-mediante PCR o hibridación in situ detectando BCR-ABL, PML-RARa • Tx-ciclofosfamida, metotrexato, citarabina ectoposido, azatioprina y antraciclina.
- 35. TX • Fase 1 de inducción= se debe lograr <5% de blastos en medula, sin blastos en sangre y >100 000 plaquetas y >1000 neutrofilos • Fase 2 postremision= para mantener la remisión. • Fase 3 continuación= durante 24 a 36 meses.
- 36. Leucemia Mieloide Crónica • Se produce por el gen quimérico BCR- ABL conocido como el cromosoma Filadelfia. • proviene de translocacion del gen BCR (22) y el gen ABL (9) generando el gen BCR-ABL • Se transcribe el ARNm y se produce la tirosincinasa BCR-ABL
- 37. Translocacion La translocacion que se produce por el intercambio de segmentos entre dos cromosomas sin pérdida de material genético se denomina translocación recíproca o equilibrada.
- 40. • BCR contiene un dominio dimerico que activa a la cinasa ABL • ABL fosforila a las proteínas RAS, AKT y JAK/STAT ( lo que les permite formar homo- o heterodímeros que se translocan al núcleo donde actuarán como activadores de la transcripción.). • Produciendo proliferación celular mieloide.
- 41. Generalidades • El gen de filadelfia se observa en 90% de los pacientes • En México es menos común la LMC que la LMA (1 /100 000) • mas frecuente en hombres (3:2) • La causa de LMC es desconocida se piensa en dosis de radiación elevadas y oncogenes
- 42. • Clasificación: • 1-tipica (Cr PH1 presente) • 2-atipica (Cr PH1 ausente) • 3-juvenil (muy agresiva) • La proliferación que resulta en un excesivo número de células mieloides en todos los estadios de maduración. • El Dx suele efectuarse con el paciente asintomático por un estudio rutinario.
- 43. • En BH: leucocitosis>30 000, anemia moderada o ausente y trombocitosis • Disminución de la Fosfatasa Alcalina leucocitica
- 44. • En los síndromes mieloproliferativos la característica es las tirosincinasa mutadas que producen la proliferación hematopoyética • La mutación se origina en células pluripotenciales. • Mutaciones como JAK 2, MPL y C-kit (mastocitosis sistémica)
- 45. Morfología de LMC • La medula es hipercelular por los precursores granulociticos. • Hay leucocitosis >100 000/mm3 y leucostasis (neutrofilos, mieloblastos, eosinofilos, mielocitos y metamielocitos y basofilos). • También hay trombosis y deposito de reticulina en la medula ósea .
- 46. Características clínicas • Afecta adultos de 50 a 60 años y la anemia leve: • 1-fatiga, hiporexia y perdida de peso • 2-molestias abdominales (dolor, plenitud y sangrado) • Esplenomegalia y hepatomegalia
- 47. • La evolución clínica de la LMC se divide en 3 fases: • 1-fase crónica(años)= hay síntomas inespecíficos, o asintomáticos al paso del tiempo evoluciona • 2-fase acelerada(6-12 meses)= hay esplenomegalia, y cels. Inmaduras en sangre y síntomas generales(fiebre,dolor oseo y perdida de peso)
- 48. • 3-fase de crisis blastica= es la evolución a Leucemia aguda y aumenta el numero de blastos en la sangre. • Los síntomas generales se intensifican y suele haber infiltrados en piel, hueso y SNC • En esta fase ya la leucemia es terminal (3 meses).
- 49. TX • 1-Imatinib (el mas eficaz) inhibe a BCR- ABL (400mg/día, oral) muy caro • 2-IFN-a (5 millones de unidades por día) con (fiebre, astenia, cefalea) • 3-Trasplante de medula ósea (el mas costoso) • A partir del Dx la supervivencia es de 4 años