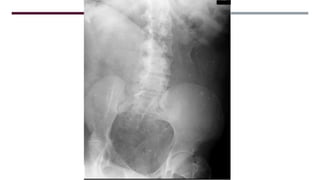
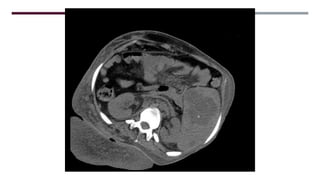
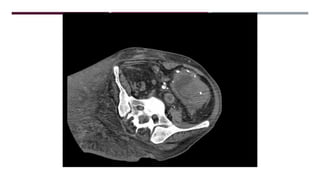
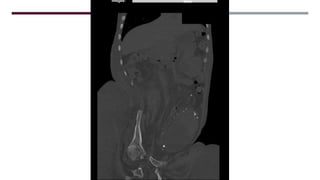
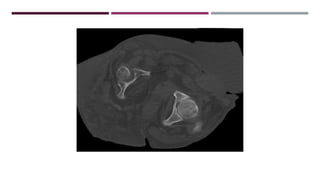
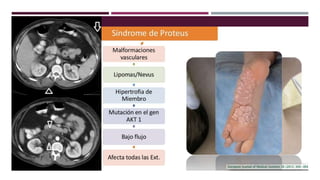
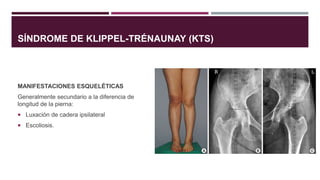
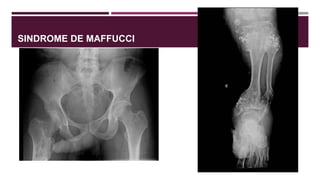
El documento describe varios síndromes congénitos raros, incluyendo el síndrome de Proteus, Klippel-Trenaunay y Maffucci, destacando sus características clínicas, radiológicas y epidemiológicas. Se mencionan anomalías específicas asociadas a cada síndrome, como crecimiento asimétrico en el síndrome de Proteus y malformaciones vasculares en el síndrome de Klippel-Trenaunay. Además, se discuten hallazgos radiográficos y la importancia de un diagnóstico clínico preciso para evitar confusiones con malignidades.