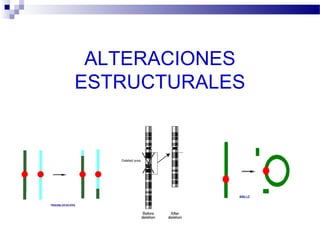
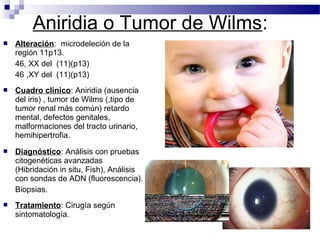
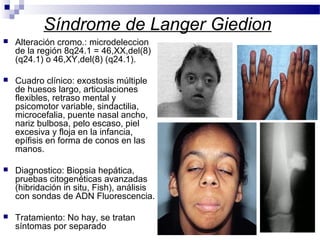
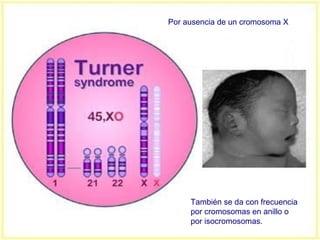
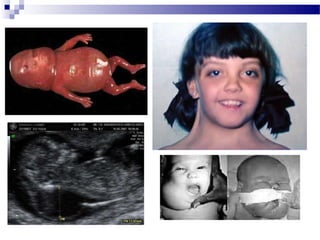
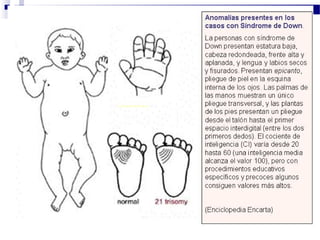
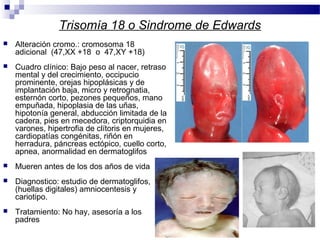
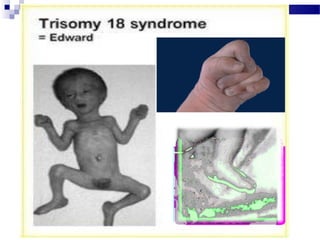
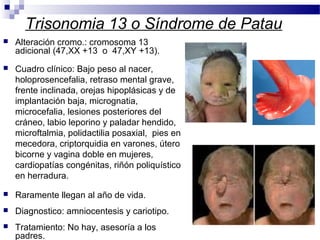
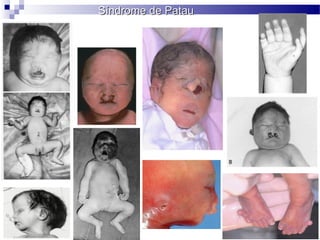
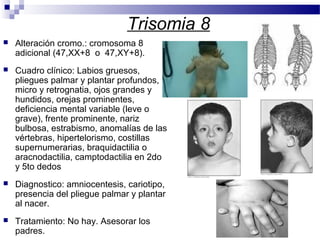
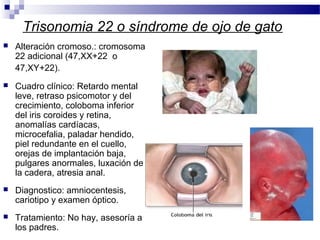
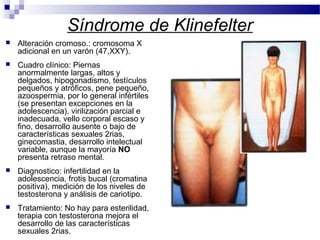
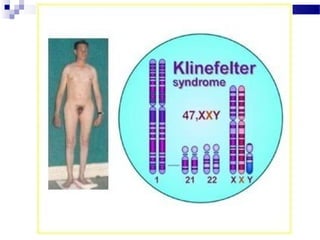
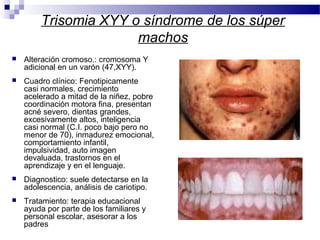
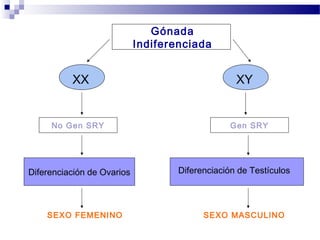
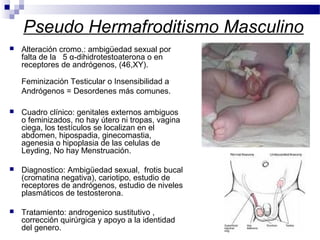
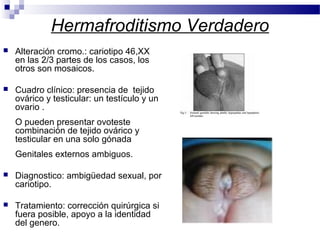
Este documento describe varios síndromes cromosómicos causados por alteraciones estructurales y numéricas de los cromosomas. Describe en detalle los síntomas clínicos, diagnóstico y tratamiento de síndromes como el de Cri du Chat, Wolf-Hirschhorn, Angelman, Prader-Willi, Williams y otros asociados a microdelecciones. También cubre síndromes por monosomías como Turner y trisonomías como Down, Edwards y Patau. El documento provee información valiosa sobre las manifestaciones fenotípicas de