FÁRMACOS ANALGÉSICOS: AINES Y OPIOIDES
•Descargar como DOCX, PDF•
3 recomendaciones•3,704 vistas

El documento resume los principales fármacos utilizados en enfermería crítica, incluyendo ácido acetilsalicílico, paracetamol, naproxeno, metamizol, diclofenaco, ibuprofeno, codeína, tramadol, dihidrocodeína, dextropropoxifeno, morfina, describiendo su farmacodinamia, farmacocinética, dosis y efectos secundarios. En total, se analizan 12 fármacos comúnmente empleados en unidades de cuidados intensivos.

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Destacado (18)
Similar a FÁRMACOS ANALGÉSICOS: AINES Y OPIOIDES
Similar a FÁRMACOS ANALGÉSICOS: AINES Y OPIOIDES (20)
Más de Centro Universitario de Ciencias de la Salud UDG
Más de Centro Universitario de Ciencias de la Salud UDG (20)
Último
Último (20)
FÁRMACOS ANALGÉSICOS: AINES Y OPIOIDES
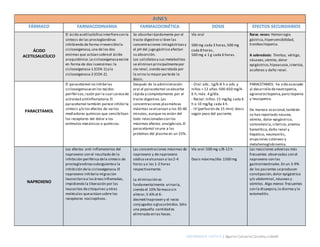
- 1. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth FÁRMACO FARMACODINAMIA FARMACOCINÉTICA DOSIS EFECTOS SECUNDARIOS ÁCIDO ACETILSALICÍLICO El ácido acetilsalicílico interfierecon la síntesis de las prostaglandinas inhibiendo de forma irreversiblela ciclooxigenasa,una de los dos enzimas que actúan sobreel ácido araquidónico.La ciclooxigenasaexiste en forma de dos isoenzimas:la ciclooxigenasa-1 (COX-1) y la ciclooxigenasa-2 (COX-2). Se absorberápidamente por el tracto digestivo si bien las concentraciones intragástricasy el pH del jugo gástrico afectan su absorción. Los salicilatosy sus metabolitos se eliminan principalmentepor vía renal, siendo excretada por la orina la mayor partede la dosis. Vía oral 500 mg cada 3 horas,500 mg cada 8 horas, 500 mg a 1 g cada 6 horas. Raras veces: Hemorragia gástrica,hipersensibilidad, trombocitopenia. A sobredosis: Tinnitus, vértigo, náuseas,vómito, dolor epigástrico,hipoacusia,ictericia, acufenos y daño renal. PARACETAMOL El paracetamol no inhibelas ciclooxigenasas en los tejidos periféricos,razón por la cual carecede actividad antiinflamatoria.El paracetamol también parece inhibirla síntesis y/o los efectos de varios mediadores químicos que sensibilizan los receptores del dolor a los estímulos mecánicos o químicos. Después de la administración oral el paracetamol seabsorbe rápida y completamente por el tracto digestivo.Las concentraciones plasmáticas máximas sealcanzan a los 30-60 minutos, aunque no están del todo relacionadascon los máximos efectos analgésicos.El paracetamol seune a las proteínas del plasma en un 25%. - Oral:ads.:1g/6-8 h o ads.y niños > 12 años:500-650 mg/4- 6 h; máx. 4 g/día. - Rectal: niños:15 mg/kg cada 6 h o 10 mg/kg cada 4 h. - IV (perfusión de 15 min): dosis según peso del paciente. PARACETAMOL ha sido asociado al desarrollo deneutropenia, agranulocitopenia,pancitopenia y leucopenia. De manera ocasional,también se han reportado náusea, vómito, dolor epigástrico, somnolencia,ictericia,anemia hemolítica,daño renal y hepático, neumonitis, erupciones cutáneas y metahemoglobinemia. NAPROXENO Los efectos anti-inflamatorios del naproxeno son el resultado de la inhibición periférica dela síntesis de prostaglandinassubsiguientea la inhibición dela ciclooxigenasa.El naproxeno inhibela migración leucocitariaa lasáreasinflamadas, impidiendo la liberación por los leucocitos decitoquinas y otras moléculas queactúan sobre los receptores nociceptivos. Las concentraciones máximas de naproxeno y de naproxeno sódico sealcanzan a las2-4 horas y a las 1-2 horas respectivamente. La eliminación es fundamentalmente urinaria, siendo el 10% fármaco sin alterar,5-6% el 6- desmetilnaproxen y el resto conjugados o glucurónidos.Sólo una pequeña cantidad es eliminada en las heces. Vía oral:500 mg c/8-12 h Dosis máxima/día:1500 mg Las reacciones adversas más frecuentes observadas con el naproxeno son las gastrointestinales.En un 3-9% de los pacientes seproducen constipación,dolor epigástrico y/o abdominal, náuseas y vómitos. Algo menos frecuentes son la dispepsia,la diarrea y la estomatitis.
- 2. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth FÁRMACO FARMACODINAMIA FARMACOCINÉTICA DOSIS EFECTOS SECUNDARIOS METAMIZOL (DIPIRONA) El metamizol actúa sobreel dolor y la fiebre reduciendo la síntesis de prostaglandinasproinflamatoriasal inhibir laactividad dela prostaglandinasintetasa. Después de su administración,el metamizol es rápidamente metabolizado por oxidación a a- metilaminoantipirina(4-MAA), 4- aminoantipirina(4-AA), 4- formilaminoantipirina (4-FAA), y 4- acetill-amino-antipirina (4-AcAA). Oral: 500 mg cada 8 horas. Vía parenteral I.M. e I.V.: Adultos y niños mayores de 12 años: 2 g por vía I.M. profunda o I.V. lenta (3 minutos) cada 8 horas. Discrasiassanguíneas (agranulocitosis,leucopenia, trombocitopenia) y choque. DICLOFENACO El mecanismo de acción del diclofenaco,como el de otros AINE, no se conoce por completo, pero parece implicar lainhibición delas vías de las ciclooxigenasas (COX-1 y COX-2) vías.El mecanismo de acción del diclofenaco también puede estar relacionado con la inhibición dela prostaglandinasintetasa. Diclofenaco seelimina a través del metabolismo y la posterior excreción urinariay la biliar del glucurónido y los conjugados de sulfato de los metabolitos.La vida media terminal de diclofenaco sin cambios es de aproximadamente2 horas.Aproximadamente el 65% de la dosis seexcreta en la orina y aproximadamente el 35% en la bilis como conjugados de diclofenaco sin cambios además de los cinco metabolitos identificados. Oral,intramuscular e intravenosa por infusión. La dosis oral va de100 a 200 mg diariamente. Adultos: Sólo aplicar las ampolletas durantedos días,y en caso necesario,sepuede proseguir con grageas de DICLOFENACO. Intramuscular: En general, la dosis es una ampolleta diariade 75 mg por vía intraglútea profunda en el cuadrante superior externo. Tracto gastrointestinal: frecuentes: dolor epigástrico,náuseas, vómitos, diarrea, calambres abdominales, dispepsia,flatulencia, anorexia. Sistema nervioso central: frecuentes: cefaleas, mareos, vértigo. Hígado: frecuentes: aumento de las transaminasasséricas IBUPROFENO Como todos los antiinflamatoriosno esteroidicos dela familia delos ácidos arilpropiónicos,el ibuprofeno inhibe la acción delas enzimas COX-1 y COX- 2. Los efectos anti-inflamatoriosdel ibuprofeno son el resultado de la inhibición periférica dela síntesis de prostaglandinassubsiguientea la inhibición dela ciclooxigenasa. El ibuprofeno se metaboliza en el hígado, dando lugar a 2 metabolitos inactivos que,junto con el ibuprofeno, se excretan por vía renal bien como tales o como metabolitos conjugados.La excreción renal es rápida y completa. Vía oral. Es posibleadministrardosis diariasdehasta 3,200 mg en dosis divididaspara el tratamiento de la artritis reumatoide y la osteoartritis, aun cuando la dosis total habitual es de 1,200 a 1,800 mg 200 a 400 mg cada 6 horas. Del 5 al 15%presentan datos de intolerancia gastrointestinal,lo más común son epigastralgias, náuseas,pirosis,sensación de plenitud en tracto gastrointestinal la pérdida oculta de sangrees infrecuente.
- 3. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth CODEÍNA La codeína es un agonista opiáceo débil en el SNC. La actividad analgésicadela codeína es debida a su conversión a la morfina.Los receptores de opiáceos están acoplados con la proteína-G (proteína de unión al nucleótido de guanina) de unión) y funcionan como moduladores, tanto positivos como negativos, de la transmisión sinápticaa través de proteínas G que activan proteínas efectoras. La codeína es bien absorbida después de la administración oral o parenteral.El inicio de acción seproduce dentro de los 10-30 minutos después de la administración intramuscular y 30-60 minutos después de la administración oral. La eliminación seproducepor vía renal como fármaco inalterado,norcodeina y morfina librey conjugada. Administración oral:Adultos:15 a 60 mg por vía oral cada 4-6 horas durante todo el día. La dosis máxima diariano debe exceder de 360 mg/24 horas. Administración parenteral: Adultos: 15 a 60 mg SC o IM cada 4-6 horas.La dosis máxima diaria no debe exceder de 360 mg / 24 horas Mareos, somnolencia, convulsiones;estreñimiento, náuseas,vómitos;prurito; erupciones cutáneas en pacientes alérgicos;confusión mental, euforia,disforia.Adosis elevadas:trastornos respiratorios,torácicos y mediastínicos,depresión respiratoria. TRAMADOL Analgésico de acción central, agonista puro no selectivo de los receptores opioides µ,delta y kappa,con mayor afinidad por los µ. En los adultos normales,la semi- vida de eliminación del tramadol y de su metabolito M1 oscilan entre las 5 y 7 horas.Tanto el fármaco nativo como sus metabolitos se eliminan principalmenteen la orina (90%) apareciendo en las heces tan solo el 10% de la dosis administrada. -Oral,formas liberación inmediata:inicial,50-100 mg; mantenimiento, 50-100 mg/6-8 h. -IM, SC, IV o en infusión:inicial, 100 mg; en la 1 a h, 50-100 mg (dolor moderado) o bien 50 mg cada 10-20 min (dolor severo) sin sobrepasar 250 mg en total; mantenimiento, 50-100 mg/6-8 h. Mareos, cefaleas,confusión, somnolencia,náuseas,vómitos, estreñimiento, sequedad bucal, sudoración,fatiga. DIHIDROCODEÍNA Acción antitusígena y analgésica central. Se absorbebien y rápidamente en el intestino. La dihidrocodeína seexcreta en leche y es capazde atravesar la placenta.Se metaboliza en el hígado. Eliminación:mediante metabolismo y posterior excreción en la orina. -Oral.En forma de dihidrocodeína bitartrato. Jarabe(12 mg/5 ml). Niños 2-5 años:3-6 mg/8 h; niños 6-12 años:6-12 mg/8 h; ads.: 12-24 mg/8 h. Gotas (10 mg/ml). Ads.: 20-30 gotas; niños:2-3 gotas por año de edad 4-5 veces/día. Molestias gastrointestinales, estreñimiento, vómitos, náuseas;fatiga,somnolencia, mareo; retención urinaria; disnea,depresión respiratoria.
- 4. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth DEXTROPROPOXIFENO El dextropropoxifeno es un analgésico narcótico queactúa sobre el sistema nervioso central. El dextropropoxifeno que es absorbido por el tracto gastrointestinal,es unido a proteínas en aproximadamente 80%, distribuyéndose ampliamente dentro del organismo en el hígado, pulmones, cerebro y riñones. Se metaboliza en el hígado. Aproximadamente de 20 a 25% de una dosis administrada,es excretada sin cambio a través de la orina siendo en mayoría recuperada como norpropoxifeno conjugado o libre.También es eliminado a través de la leche materna en pequeñas cantidades. La vía de administración de DEXTROPROPOXIFENO es oral. La dosis en adultos que se recomienda es de 65 mg cada 4 horas,sin exceder de 360 mg/día. Náusea, vértigo, boca seca, constipación,confusión, bradicardia,palpitaciones, hipotensión ortostática, alteraciones emocionales, disminución dela libido, alucinaciones y euforia a dosis alta.
- 5. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth MORFINA Analgésico agonista delos receptores opiáceos µ,y en menor grado los kappa,en el SNC. Se absorbemuy bien por el intestino y, por vía rectal,su absorción es incluso más rápida. La morfina se metaboliza fundamentalmente en el hígado mediante las enzimas del citocromo P450 2D6, pero también se metaboliza parcialmente en el cerebro y los riñones. La morfina se elimina en forma de los conjugados anteriores por vía urinaria y biliar. Sol. inyectablede morfina hidrocloruro al 1%o 2%: - Vía SC o IM: ads.:5-20 mg/4 h. - Iny. IV lenta: ads.: 2,5-15 mg en 4-5 min; en IAM se pueden administrar dosisen aumento (1-3 mg) hasta cada 5 min. - Perfus. IV continua:ads.: inicial 0,8-10 mg/h; mantenimiento, 0,8-80 mg/h; hasta 440 mg/h en exacerbaciones. - Epidural lumbar:sólo ads.:5 mg. - Intratecal lumbar:sólo ads.: 0,2-1 mg/24 h. Confusión,insomnio, alteraciones del pensamiento, cefalea,contracciones musculares involuntarias, somnolencia,mareos, broncoespasmo,disminución de la tos, dolor abdominal, anorexia,estreñimiento, sequedad de boca,dispepsia, nauseas,vómitos,hiperhidrosis, rash,astenia,prurito.Depresión respiratoria.Retención urinaria (más frecuente vía epidural o intratecal). FENTANILO De la misma manera que la morfina,el fentanilo es un fuerte agonista de los receptores opiáceos µy kappa. El fentanilo experimenta un metabolismo del hígado a través de la oxidación a norfentanilo seguida a continuación,por la hidrólisisa 4-N-anilinopiperidina y ácido propiónico.Los metabolitos y el fármaco inalterado seexcretan en la orina,lo cual puede tomar varios días. Premedicación,IM: 0,05-0,10 mg. Inducción,IV: 0,05-0,10 mg inicialmente; repetir cada 2-3 min hasta conseguir efecto deseado. Mantenimiento, IVo -- IM: 0,025-0,05 mg según tensión arterial durantela intervención. Postoperatorio, IM: 0,05-0,10 mg (sala de recuperación). Depresión respiratoria,apnea, rigidezmuscular,bradicardia, vértigo, visión borrosa,cambios de humor, náuseas,vómitos, laringoespasmos,diaforesis, espasmo del esfínter de Oddi, hipotensión. METADONA Agonista opiáceo puro de origen sintético con potencia ligeramente superior a la morfina,mayor duración de acción y menor efecto euforizante. Presenta afinidad y marcada actividad en los receptores µ. La metadona posee una fasede distribución,con una vida media de 2-3 horas y una vida media de eliminación de15-60 horas. La eliminación fecal representa la mayoría de la excreción de la metadona. - Dolor: oral y SC: 5-10 mg/dosis según intensidad. -Oral:inicial:20-30 mg/día, según respuesta aumentar hasta 40-60 mg/día en 1 a 2 sem. Mantenimiento: 60-100 mg/día, alcanzándosecon incrementos sucesivos semanales de10 mg/día. Aturdimiento, mareo, sedación, náuseas,vómitos,sudoración, euforia,disforia,debilidad, cefalea,insomnio,agitación, desorientación,alteraciones visuales,boca seca,anorexia, estreñimiento, espasmo del tracto biliar,rubor cutáneo, bradicardia,palpitaciones, desmayo, síncope, retención o tenesmo urinario,efecto antidiurético,disminución de la libido y/o potencia sexual, prurito,urticaria,exantema cutáneo, edema.
- 6. ENFERMERÍA CRÍTICA | Aguilar Calvario Carolina Lizbeth OPIOIDES FUERTES OXICODONA Agonista puro opioidecon afinidad por receptores opiáceos µ, kappa y delta, con efecto analgésico,ansiolítico y sedante. La farmacocinética dela oxicodona es lineal dentro del rango de dosis de5 a 15 mg. En los comprimidos deoxicodona de liberación sostenina,la liberación del analgésico es independientee del pH y la biodisponibilidad es del 60% al 87%, si bien es del 100% en comparación con los comprimidos de liberación inmediata. La oxicodona y sus metabolitos se excretan principalmentepor vía renal. -Oral.Ads. > 20 años,inicial: forma retardada,10 mg/12 h; formas de liberación inmediata, 5 mg/4-6 h; titular con incrementos del 25-50%. -I.V. (en bolus):1-10 mg lentamente durante 1-2 min, no administrar con una frecuencia > 4 h. I.V. (perfusión):inicial 2 mg/h. I.V. (analgesia controlada por el paciente PCA): en bolus 0,03 mg/kg en un tiempo mín. de 5 min. -SC (en bolus):inicial 5 mg, repetir si necesario a intervalos de 4 h. SC (perfusión):inicial 7,5 mg/día. Estreñimiento, náuseas, vómitos, dolor abdominal, anorexia,diarrea,boca seca, dispepsia,flatulencia;edema; somnolencia,mareos,sueños anómalos,ansiedad,confusión, depresión, insomnio, alucinaciones,nerviosismo, debilidad,astenia,cefalea; vasodilatación,hipotensión ortostática;disnea, broncoespasmo;prurito,rash, sudoración;trastornos urinarios; fiebre, escalofríos. BUPRENORFINA La buprenorfina es un potente agonista de los receptores opiáceos µ. Los receptores opiáceos incluyen los µ(mu), k (kappa),y d (delta), todos ellos acoplados a los receptores para la proteína G y actuando como moduladores,tanto positivos como negativos de la transmisión sinápticaquetiene lugar a través de estas proteínas. El fármaco semetaboliza parcialmente por N- desalquilación y conjugación.Se elimina principalmentepor vía fecal,recuperándose en las heces aproximadamente el 70% de la dosis administrada.Un 20% se elimina por vía renal en forma de conjugados. -Administración oral: Adultos: se administrará inicialmente un comprimido cada 8 horas,que podrá aumentarse, según la intensidad de dolor,a 1-2 comprimidos cada 6-8 horas. -Administración parenteral: Adultos: se administrarápor vía intramuscularo intravenosa una o dos ampollas deBuprenorfina de 0,3 mg según la intensidad del dolor.Esta dosis puedeser repetida, si es necesario,cada 6- 8 horas. Puede aparecer somnolencia, fácilmente reversible, especialmente en el postoperatorio.Ocasionalmente se ha observado una ligera euforia.Puede producirse depresión respiratoria,náuseas, vómitos, vértigos y sudoración en algunos pacientes ambulatorios.