Descargado 16 veces




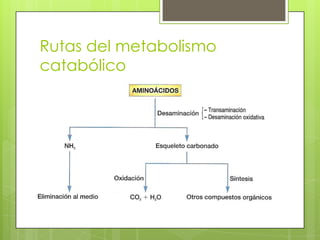


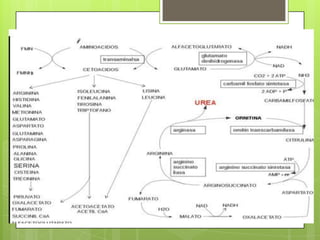
















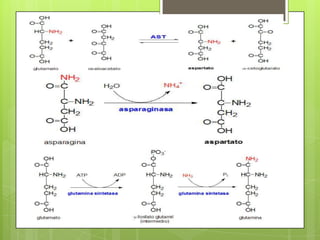








El documento describe las rutas metabólicas de las proteínas en el cuerpo. 1) Las proteínas se digieren en el estómago y intestino delgado por enzimas como la tripsina, y los aminoácidos resultantes se absorben en el intestino delgado acoplados al transporte de sodio. 2) En el hígado y otros tejidos, los aminoácidos se descomponen a través de reacciones como la transaminación, desaminación y descarboxilación para producir compuestos como el piruvato y el acetil-CoA,











































































































